
Sitges, 17, 18 y 19 de mayo 2007
COMUNICACIONES ORALESSARCOMAS DE PARTES BLANDAS NO RABDOMIOSARCOMAS EN ADOLESCENTESN. Pardo1, M. Melo2, J. Almazán3, M. Torrent1, I. Badell1, P. Abad2 y S. Brió1
1G.I.C.O.P. Hospital Sant Pau. 2Hospital Parc Taulí. Sabadell. 3Hospital Germans Trias i Pujol. Badalona. Barcelona. España.
Introducción: Bajo el término “sarcomas de partes blandas” (SPB) se agrupa un conjunto de entidades malignas que se originan a partir del tejido mesenquimal primitivo, y de ellas los rabdomiosarcomas suponen el 60 % en niños menores de 5 años. No obstante, en adolescentes esta tasa disminuye para dar paso al grupo de SPB no rabdomiosarcomas, que asciende a cifras superiores al 70 %, y con incidencia de alrededor de 10 casos por millón de población entre 10 y 20 años.
Material y métodos: entre enero de 1992 y enero de 2007 hemos diagnosticado 16 casos de SPB no RMS en pacientes de edades comprendidas entre 10 y 20 años. Los diagnósticos fueron: 7 sarcomas sinoviales (SS), 2 fibrosarcomas (FS), 2 sarcomas miofibroblásticos (SMF), 2 fibromatosis agresivas (FA), 1 histiocitoma fibroso maligno (HFM), 1 tumor desmoplásico (TD) y 1 sarcoma epitelioide (SE). La media de edad fue de 13 años. Todos los pacientes fueron candidatos a cirugía, que en 11 fue radical. En 12 pacientes se administró quimioterapia según protocolo SIOP y 8 recibieron radioterapia.
Evolución: De los sarcomas sinoviales, 1 acabó falleciendo a los 7 años del diagnóstico inicial tras recidiva y tratamientos de segunda línea, los otros 6 están vivos, 1 con enfermedad y en tratamiento y 5 con supervivencias libres de enfermedad de 15, 24, 24, 81 y 128 meses. De los 2 fibrosarcomas, ambos consiguieron remisión completa, pero uno recidivó localmente a los 27 meses del diagnóstico y, pese a la amputación de la extremidad afectada, la enfermedad progresó ganglionarmente y llegó a metástasis pulmonares que ocasionaron la muerte a 75 meses de su inicio. Los 2 pacientes con fibromatosis agresivas están vivos con enfermedad a 37 y 26 meses del diagnóstico, han sido objeto de repetidas cirugías y en el momento actual han iniciado tratamiento quimioterápico. Los 2 sarcomas miofibroblásticos están vivos sin enfermedad a 25 y 128 meses. Los otros 3 pacientes están vivos sin enfermedad y fuera de tratamiento a 25 meses el HFM, 28 meses el tumor desmoplásico y 32 meses, respectivamente.
Resultados: La supervivencia global es del 87 % y la libre de enfermedad, del 68 %. Aunque los resultados son aceptables, creemos que podrían disminuir dado el alto riesgo de recidivas locales y metastásicas a largo plazo que estos tumores plantean.
REVISIÓN DE LA CALIDAD DE VIDA EN ADOLESCENTES SUPERVIVIENTES DE CÁNCERN. Martínez Ezquerra1, P. Garrido Valtierra1, A. Echebarría Barona1, I. Astigarraga Aguirre1, A. Fernández-Teijeiro Álvarez1, S. Rodríguez Tejedor2 y A. Navajas Gutiérrez1
1Unidad de Oncología Pediátrica, Hospital de Cruces. Baracaldo. Vizcaya. 2Servicio de Documentación Médica, Hospital de Cruces. Baracaldo. Vizcaya. España.
Objetivos: Conocer las desviaciones de la normalidad e integración escolar y social de un grupo de adolescentes supervivientes de cáncer tratados en nuestra unidad de oncología pediátrica.
Material y métodos: Estudio retrospectivo de pacientes tratados en los últimos 10 años. Los criterios de inclusión fueron los siguientes: pacientes diagnosticados desde 1996 hasta 2006, edad actual > 10 años y remisión de la enfermedad en el momento del estudio. Se excluyeron fallecidos y tumores benignos. Posteriormente, se diseñó una encuesta de calidad de vida modificada de HUI y PEDQOL. Se realizó entrevista personal o telefónica. Para el análisis estadístico de los datos se utilizó el programa SPSS para Windows versión 14.0.
Resultados: De los 79 adolescentes supervivientes entrevistados, el 54 % (n = 43) eran varones. La edad media fue de 180 meses (± 35,75 DE) y la edad media al diagnóstico fue de 71,44 meses (± 52,14 DE). La supervivencia media libre de enfermedad fue de 94,46 meses (± 57,74 DE). El 8,9 % presentaron recidiva. El tipo diagnóstico más frecuente correspondió a LLA (25 %), tumores del sistema nervioso central (20,2 %) y neuroblastoma (15,2 %). El 35,4 % se trataron con cirugía y quimioterapia, el 27 % con quimioterapia y el 16,5 % solamente con cirugía. El 55 % de los casos no presentaban ninguna secuela postratamiento. Las secuelas psicológico-psiquiátricas y sensoriales fueron del 7,7 %; las motoras, del 6,4 %, y las neurológicas y endocrinas del 5,1 %, respectivamente. El 97,5 % de los adolescentes estaban escolarizados y el 32,9 % había repetido curso. Respecto a las relaciones sociales, el 87,5 % refería no tener problemas para relacionarse. El 50,5 % realizaba deporte tanto escolar como extraescolar, con una frecuencia media de 2 veces por semana. El 20 % de los entrevistados referían ser consumidores de alcohol de forma esporádica y el 3,8 % fumaba. El 98,7 % de los pacientes eran autónomos para desenvolverse en la vida diaria. El 94,93 % se consideraba contento y adaptado. El rechazo a colaborar con la entrevista fue del 1,3 %.
Conclusión: A pesar de haber presentado un proceso oncológico en la infancia observamos que la mayoría de los supervivientes han sabido adaptarse de forma adecuada a la etapa de la adolescencia, con hábitos de vida semejantes al resto de las personas de su misma edad. El mayor porcentaje de secuelas correspondió al grupo de tumores cerebrales en los campos neurocognitivo, psicológico y sensorial.
INTEGRACIÓN DE LA PET EN EL ESTUDIO Y TRATAMIENTO DEL LINFOMA DE HODGKINA. Herrero Hernández, O. Escobosa Sánchez, R. Calvo Medina y T. Acha García
Unidad de Oncología Pediátrica. Hospital Materno-Infantil Carlos-Haya. Málaga. España.
Objetivo: En los últimos años se ha incrementado el uso de las imágenes funcionales de la PET en el linfoma de Hodgkin (LH), para aumentar la especificidad en detectar tejido vital frente a residuo fibrótico-necrótico. En el estudio EuroNet-PHL.C-1 la PET estará formalmente integrada en el estadiaje y evaluación de la respuesta. Se presentan los resultados en algunos de nuestros pacientes.
Material y métodos: Se analizan un total de 10 pacientes diagnosticados de LH en nuestra unidad desde enero de 2003 hasta marzo de 2007. Se han evaluado según recomendaciones del protocolo actual del grupo SEOP (EH-SEOP.003), excluyéndose recidivas y progresiones. Se integran resultados de la PET realizada en nuestros pacientes: al diagnóstico (7/10), en la evaluación de la remisión al cuarto ciclo (6/10) y al final del tratamiento (7/10). Equipamiento: tomógrafo PET Siemens ECAT-EXACT 47. Dosis: 6 MBq/kg (mínimo 37 MBq) de 18F-FDG (fluordeoxiglucosa) intravenoso.
Resultados: Las edades en el momento del diagnóstico iban de 6 a 15 años. Histología: 6 de 10 esclerosis nodular, 2 predominio linfocítico, 1 deplección linfocítica, 1 celularidad mixta. Todos están vivos con tiempo de seguimiento de 1 a 48 meses. Han recibido radioterapia 5 de 10. En el diagnóstico 2 de 7 presentaron menor estadiaje por PET, pero no se determinaron cambios en el grupo terapéutico. En la evaluación de la remisión al cuarto ciclo 5 de 6 pacientes presentaban RCNC con pruebas convencionales y RC con PET, 1 de 6 en BRP con PET negativo. Al final del tratamiento, 6 de 7 pacientes estaban en RCNC con PET negativa y 1 de 7 en RC con PET negativa.
Conclusiones:1) En nuestros pacientes la utilización de la PET al diagnóstico no ha supuesto ningún cambio de grupo terapéutico a pesar del menor estadiaje en 2 de ellos (regiones discordantes negativas en PET y positivas en TC/RM). 2) No se han considerado en nuestros pacientes regiones discordantes positivas en PET y negativas en TC/RM. 3) En la valoración de la respuesta al cuarto ciclo de quimioterapia y al final del tratamiento según protocolo EH-seop.003, la PET ha sido resolutiva en la determinación de la remisión completa de todos nuestros pacientes.
CRIOPRESERVACIÓN DE TEJIDO OVÁRICO EN NIÑAS Y ADOLESCENTES CON CÁNCERO. Cruz1, E. Martínez2, J. Callejo2, J. Mora1, A. Parareda1, C. De Torres1, S. Segura1 y A. Laguna1
Servicios de 1Oncología y 2Ginecología. Hospital Universitario Sant Joan de Déu. Universidad de Barcelona. España.
Objetivos: Las dosis altas de alquilantes, la irradiación corporal total y la irradiación abdominopélvica se asocian con un alto riesgo de lesión en la función ovárica, incluyendo la pérdida de fertilidad. En las niñas y adolescentes la única forma de preservar la función gonadal y la fertilidad es la criopreservación de tejido ovárico (CO). Este estudio pretende comunicar nuestra experiencia en la CO.
Pacientes y métodos: Se ofrece la posibilidad de CO a todas las pacientes en las que el tratamiento previsto supone un alto riesgo de fallo ovárico. La paciente y sus padres son informados del procedimiento a la hora de firmar el consentimiento. El procedimiento fue aprobado por el comité ético del hospital. La recogida de tejido ovárico se realiza siempre que es posible antes de comenzar el tratamiento esterilizante. La CO se efectúa mediante laparoscopia, con tres puntos de abordaje En todos los casos se recoge todo un ovario. Para la criopreservación se fragmenta la cortical del ovario y dichos fragmentos se congelan y almacenan. Una porción del ovario se remite para análisis histológico.
Resultados: Desde noviembre del año 2000 se han sometido a CO 23 pacientes. Los diagnósticos fueron linfoma de Hodgkin (n = 8), sarcoma de Ewing (n = 7), osteosarcoma (n = 5), astrocitoma de alto grado (n = 1), linfoma linfoblástico (n = 1), y tumor rabdoide extrarrenal (n = 1). Los tratamientos de riesgo fueron autotrasplante de médula (n = 3), quimioterapia con altas dosis de alquilantes (n = 18) y radioterapia pélvica (n = 2). La edad media en el momento de la preservación fue de 14,4años (rango 10 a 18 años). En 10 casos (43 %) la CO se efectuó tras haber recibido tratamiento quimioterápico, porque la gravedad de la enfermedad obligaba al inicio de la quimioterapia (n = 4), por efectuarse en recidiva (n = 3) o por demora en la decisión familiar o razones administrativas (n = 3). No existieron complicaciones quirúrgicas, salvo un caso de infección de la herida. En la mayoría de los casos el ovario preservado fue el derecho. En ningún caso se observó tumor en el fragmento ovárico analizado. Un total de 3 pacientes han fallecido por progresión de la enfermedad (13 %).
Conclusiones: La CO puede ofrecerse sistemáticamente a las pacientes con tratamientos antineoplásicos de alto riesgo gonadal. Nuestra experiencia y la de otros autores nos motiva a que sea también ofrecida a las niñas prepuberales.
POLIPOSIS FAMILIAR HEREDITARIA EN LOS NIÑOS Y ADOLESCENTES: VALOR DEL ESTUDIO GENÉTICO E INDICACIONES DE LA COLECTOMÍA PROFILÁCTICAJ. Balaguer1,2, S. Oltra3 , A. Segura4 , M. García4, A. Pereda5 y V. Castel1
1Unidad de Oncología Pediátrica. 2Fundación para la Investigación. 3Unidad Genética. 4Unidad del Consejo Genético Cáncer. 5Unidad de Gastroenterología Pediátrica. Hospital Universitario La Fe. Valencia. España.
Introducción: Mutaciones germinales en el gen supresor de tumores APC (Adenomatous Polyposis Coli) son responsables de la mayoría de los casos de poliposis adenomatosa familiar (FAP). Es una patología de herencia autosómica dominante, con una penetrancia del 95 %, causante del 1 % de los tumores colorrectales. El diagnóstico clínico se basa en la presencia de múltiples pólipos adenomatosos en colon y recto que pueden causar un cáncer colorrectal, por lo que se consideran lesiones premalignas que necesitan de cirugía profiláctica. En la mayoría ocurre hacia los 40 años, pero hasta el 15 % puede ocurrir en la adolescencia. Los estudios genéticos han permitido la identificación de portadores, ofreciendo a estas familias un diagnóstico presintomático precoz y un correcto asesoramiento genético, tanto en los portadores como en los no portadores. La forma grave se ha correlacionado con la afectación de los codones 1250 a 1464 del exón 15 del gen APC, y las formas atenuadas con la afectación distal.
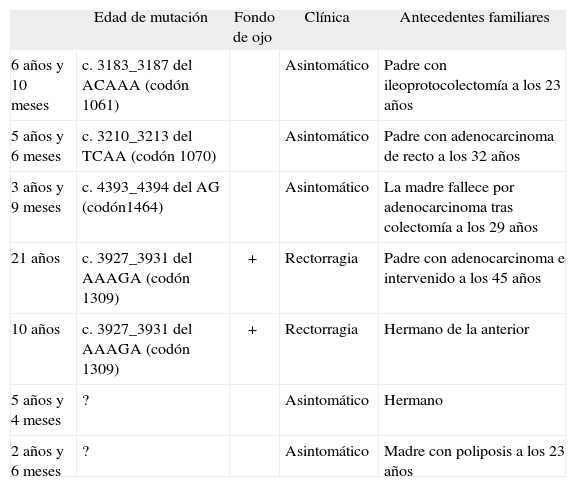
Pacientes y método: Revisión retrospectiva de las familias PAF del registro de la Unidad de Consejo Genético del Cáncer (UCGC). De 26 familias con 56 portadores, 4 familias tienen 5 portadores diagnosticados en edad pediátrica. Otras 2 familias con 3 pacientes se han diagnosticado en la consulta por clínica y 2 pacientes se han identificado a partir del estudio familiar. El estudio genético se realizó mediante amplificación por PCR de las regiones exónicas y las intrónicas colindantes del gen APC. Análisis mediante SSCP's/Heteroduplex con secuenciación directa en ambos sentidos en los casos que se observaron patrones anómalos. También se buscaron grandes reordenamientos por la técnica de MLPA.
Resultados (tabla 1):
TABLA 1.
| Edad de mutación | Fondo de ojo | Clínica | Antecedentes familiares | |
| 6 años y 10 meses | c. 3183_3187 del ACAAA (codón 1061) | Asintomático | Padre con ileoprotocolectomía a los 23 años | |
| 5 años y 6 meses | c. 3210_3213 del TCAA (codón 1070) | Asintomático | Padre con adenocarcinoma de recto a los 32 años | |
| 3 años y 9 meses | c. 4393_4394 del AG (codón1464) | Asintomático | La madre fallece por adenocarcinoma tras colectomía a los 29 años | |
| 21 años | c. 3927_3931 del AAAGA (codón 1309) | + | Rectorragia | Padre con adenocarcinoma e intervenido a los 45 años |
| 10 años | c. 3927_3931 del AAAGA (codón 1309) | + | Rectorragia | Hermano de la anterior |
| 5 años y 4 meses | ? | Asintomático | Hermano | |
| 2 años y 6 meses | ? | Asintomático | Madre con poliposis a los 23 años |
Conclusiones: El estudio de mutaciones del gen APC para identificar portadores debe considerarse una prueba diagnóstica habitual en los niños pertenecientes a familias de riesgo, así como en niños con clínica e historia compatible. En nuestra serie, todos son PAF clásica, 2 familias se encuadrarían dentro de la PAF grave. En una de ellas existen 2 hermanos portadores de 21 y 10 años. La chica de 21 años está pendiente de colectomía profiláctica por degeneración adenomatosa de sus pólipos. El riesgo de malignización es a medio plazo, por lo que la adolescencia es un período en el que es necesario un seguimiento estricto, y en caso de clínica se debe realizar una colonoscopia diagnóstica y/o biopsia de pólipos. La colectomía profiláctica debe realizarse en el momento óptimo, antes de la malignización de los pólipos, pero permitiendo un adecuado desarrollo pondoestatural prepuberal dentro de lo posible.
QUIMIOTERAPIA CITORREDUCTORA Y RADIOTERAPIA EXTERNA EN RETINOBLASTOMA AVANZADOC. Sábado1, M. Arguis2, N. Martín3, L. Gros1, J.L. Dapena1, S. Gallego1, J. Giralt2 y J. Sánchez de Toledo1
1Servicio de Oncología-Hematología Pediátrica. 2Servicio de Radioterapia. 3Servicio de Oftalmología. Hospital Vall d'Hebron. Barcelona. España.
Objetivos: Valorar el efecto del tratamiento con quimioterapia citorreductora y radioterapia externa sobre la preservación ocular, el control tumoral y la tasa de complicaciones en pacientes con retinoblastoma avanzado.
Métodos: Los pacientes recibieron tratamiento inicial con quimioterapia y radioterapia externa (RT) desde enero de 1996 hasta marzo de 2006. El esquema de quimioterapia consistió en 6 ciclos de carboplatino, vincristina y etopósido administrados cada 4 semanas. Se administró RT conformada 3-D con una fuente de fotones de 6 MV en una dosis total de 40 Gy en 20 fracciones. La toxicidad tardía fue evaluada usando los criterios estandarizados de toxicidad. El seguimiento se efectuó mediante examen de fondo de ojo realizado con anestesia general.
Resultados: Se evaluaron 24 pacientes con un total de 37 ojos afectados. Un total de 3 pacientes (12 %) presentaban historia familiar. La edad media en el momento del diagnóstico era de 17 meses (rango 1-84 meses); 13 pacientes (52 %) presentaban tumores bilaterales y 12, tumores unilaterales. Según la clasificación de Reese-Ellsworth, 7 eran tumores del grupo III, 4 del grupo IV y 26 del grupo V. Un total de 10 pacientes presentaban siembras vítreas al diagnóstico; 3 de los 13 pacientes con retinoblastoma bilateral fueron sometidos a enucleación unilateral como tratamiento inicial. Con una mediana de seguimiento de 60 meses, la tasa de recurrencia local fue del 28 %, sin que se observaran metástasis a distancia. La tasa de preservación ocular tras quimiorradioterapia fue del 77 %. La supervivencia global fue del 100 %. Las complicaciones tardías incluyeron cataratas (40 %), xeroftalmía (6 %) y retinopatía (2 %). Un paciente desarrolló una neoplasia secundaria en el campo de irradiación (osteosarcoma).
Conclusiones: En el retinoblastoma avanzado la quimioterapia citorreductora, junto con la irradiación externa, consigue tasas elevadas de preservación ocular y de control tumoral. El riesgo de complicaciones tardías es moderado.
PÓSTERSENFERMEDAD DE HODGKIN EN ADOLESCENTES. A PROPÓSITO DE DOS CASOS DE TÓRPIDA EVOLUCIÓNC. Navarro Moreno, E. Pérez Santaolalla, I. Ruiz Alcántara y A. Verdeguer Miralles
Oncología Pediátrica. Hospital Universitario La Fe. Valencia. España.
Introducción: La enfermedad de Hodgkin (EH) es un linfoma maligno constituido por la proliferación clonal de linfocitos B acompañados de células inflamatorias y reactivas. Es un tumor poco frecuente en niños y tiene una tasa de curación elevada. En pediatría, la mayoría de los casos se diagnostican por encima de los 12 años y es en este subgrupo la enfermedad presenta un curso más abigarrado. Presentamos 2 casos clínicos de EH diagnosticados en adolescentes.
Caso clínico 1: Paciente diagnosticada a los 13 años de EH tipo esclerosis nodular estadio IIB de localización supradiafragmática. Se inicia tratamiento quimioterápico según protocolo SEOP EH-2/97 sin lograrse remisión completa (RC), por lo que se administra radioterapia, tras la que persiste lesión residual mediastínica. Reactivación de la enfermedad a los 10 meses del diagnóstico, que se trata con una combinación de QMT, RT y TPH autólogo, alcanzándose RC documentada por PET. A los 15 meses pos-TPH, se diagnostica nueva recidiva, y resulta ineficaz el tratamiento QMT de rescate. Se realiza TPH alogénico de hermana HLA idéntica previo acondicionamiento de intensidad reducida.
Caso clínico 2: Paciente diagnosticada a los 12 años de EH esclerosis nodular de predominio linfocítico en estadio IV. Recibe tratamiento quimioterápico según protocolo EH-2/97, sin alcanzar la RC. Una segunda línea de QMT resulta también inefectiva. Se procede entonces a TPH autólogo (acondicionamiento con BEAM) y posterior irradiación de los restos tumorales. Desde entonces ha presentado hasta un total de 4 recidivas, tratadas con diversas combinaciones de QMT con buena respuesta clínica que ha permitido períodos libres de tratamiento, pero persiste un resto tumoral pulmonar. Desde hace unos meses, se objetiva nueva progresión de la enfermedad en la zona infradiafragmática, por lo que se ha reiniciado de nuevo quimioterapia con buena respuesta.
Comentarios y conclusiones: Aunque es conocido el diferente comportamiento de las recidivas en la EH, con una mayor respuesta que en otras neoplasias, la evolución de estas dos pacientes es especialmente tórpida. En los casos presentados, a pesar de los múltiples e intensivos tratamientos, ambas pacientes siguen un régimen de vida normal.
CARCINOMA NASOFARÍNGEO EN PEDIATRÍA: PRESENTACIÓN DE DOS CASOSA. Herrero Hernández, R. Calvo Medina, O. Escobosa Sánchez y T. Acha García
Unidad de Oncología Pediátrica. Hospital Materno-Infantil Carlos-Haya. Málaga. España.
Introducción: El carcinoma nasofaríngeo es un tipo de tumor raro en la edad pediátrica, más frecuente en varones de 10 a 14 años. En nuestro país se diagnostica especialmente en pacientes procedentes del Magreb (área endémica), y los tipos II y III se asocian a la infección por VEB. Los IgG e IgA frente a Ag cápside viral presentan correlación con la masa tumoral y son útiles para valorar la efectividad del tratamiento y las posibles recidivas. Altas dosis de radioterapia acompañada de quimioterapia consiguen supervivencia global del 77 % a los 5 años.
Material y métodos: Presentamos dos casos asistidos en nuestra unidad procedentes de Marruecos. Ambos han sido tratados según protocolo NPC-2003-GPOH para estadio III y grupo de alto riesgo, con 3 ciclos de cisplatino (100 mg/m2/día, D1) y 5-FU (1.000 mg/m2/día, D1-D5) seguidos de radioterapia (59,4 Gy tumor primario y 50 Gy adenopatías cervicales) simultánea con 2 ciclos de cisplatino (20 mg/m2/día D1-D3). Caso 1: varón de 11 años con tumoración cervical izquierda progresiva de 4 meses de evolución, acompañada de síntomas constitucionales, epistaxis, cefalea e hipoacusia. A la exploración destaca adenopatías laterocervicales izquierdas duras de 3–4 cm, no dolorosas, adheridas. Serología VEB: IgG VCA (630), IgM VCA(−), PCR VEB (+). Ecografía y TC de la región cervical: masa faríngea que afecta a la totalidad del anillo de Waldeyer, con grandes adenopatías laterocervicales. Biopsia cervical: metástasis de carcinoma nasofaríngeo indiferenciado. Tras tratamiento con el protocolo referido está vivo libre de eventos a los 4 años de seguimiento, con bajos títulos de anticuerpos anti-VEB. Presenta secuelas dentales. Caso 2: varón de 13 años con episodios de hemoptisis, febrícula, voz nasal y poliadenopatías cervicales bilaterales de 2 meses de evolución. En la exploración: adenopatías cervicales duras, adheridas a planos profundos. Serología VEB: IgG VCA (590) IGM(−), PCR a VEB (+). Ecografía, TC y RM: adenopatías cervicales de aspecto tumoral y masa retrofaríngea de 5,5 × 4 cm que penetra en fosas nasales. Biopsia ganglionar: metástasis de carcinoma nasofaríngeo indiferenciado. Tras tratamiento con quimioterapia y radioterapia sigue vivo libre de eventos a los 3 años de seguimiento. Presenta secuelas dentales e hipotiroidismo posradioterapia.
Conclusiones: La epistaxis recidivante en pacientes de áreas endémicas debe alertar del carcinoma nasofaríngeo. Altas dosis de radioterapia combinada con poliquimioterapia es un tratamiento efectivo. Dada su asociación al VEB, son marcadores útiles de respuesta al tratamiento y recidivas los anticuerpos frente a cápside viral.
¿RECIDIVA TUMORAL O TERCERA MALIGNIDAD?M. Delgado, J.L. Dapena, S. Gallego, L. Gros, C. Sábado y J. Sánchez de Toledo
Servicio de Oncología-Hematología Pediátrica. Hospital Vall d'Hebron. Barcelona. España.
Muy reconocido es el alto riesgo de segundas malignidades observado en pacientes con retinoblastoma hereditario (RBH). La alta tasa de curación observada en el RBH asociado al tratamiento con radioterapia externa (RTE) permite ahora que los pacientes alcancen la edad adulta y, con ello, la aparición de segundos tumores.
Objetivo: Se presenta el caso de adolescente tratada de retinoblastoma hereditario (RBH) en su infancia que 13 años después presenta una segunda neoplasia y un tercer evento tumoral 3 años después.
Caso clínico: Adolescente de 17 años, tratada de RBH a los 3 meses de edad mediante enucleación de ojo izquierdo y radioterapia externa (RTE) sobre el ojo derecho (OD) (33 Gy). Presenta recidiva tumoral temprana en el OD que requiere tratamiento con quimioterapia, RTE (20 Gy) y crioterapia. A los 13 años edad y de su primer inicio tumoral presenta una segunda neoplasia con clínica de epistaxis y obstrucción nasal derecha. Se le diagnostica de sarcoma osteogénico (SO) de seno maxilar derecho, fosa nasal derecha, celdas etmoidales y reborde alveolar maxilar superior derecho. Es tratada con quimioterapia neoadyudante con vincristina, ifosfamida, actinomicina D, metotrexato y cisplatino y cirugía tumoral con resección macroscópica total. Presenta recidiva temprana de SO en vómer y maxilar derecho al finalizar quimioterapia inicial, la cual es tratada con cirugía, RTE y quimioterapia (un ciclo ICE y cisplatino) con remisión tumoral completa. Tres años después presenta nuevamente clínica de epistaxis y obstrucción nasal y mediante RM y TC se le diagnostica SO de huesos esfenoides y etmoides con metástasis pulmonares. Recibe tratamiento con protocolo de rescate consistente en metrotexato (12 g/m2), ifosfamida (14 g/m2) y cisplatino (90 mg/m2), al que se agrega intervención quirúrgica y radiocirugía sobre el área tumoral. Actualmente, la paciente se encuentra en remisión completa tras 3 meses de finalizado el tratamiento. Como efectos adversos del tratamiento recibido asocia hipoacusia bilateral importante, hipoplasia en la hemicara derecha, disminución de agudeza visual en el ojo derecho, nefrotoxicidad y aparición de segunda neoplasia. Puesto que el origen de este tercer evento neoplásico parece ser un hueso distinto al del primer sarcoma osteogénico, se plantea la posibilidad de un tercer evento neoplásico en una base de predisposición genética en lugar de una recidiva tumoral.
Conclusiones: Las diferentes modalidades terapéuticas empleadas en esta paciente ilustran de manera clara la evolución en el tratamiento del retinoblastoma. La sobrevida alcanzada por estos pacientes en la actualidad ha permitido la aparición de efectos adversos tardíos de la quimioterapia o radioterapia. Con el progreso alcanzado en las diferentes modalidades de tratamiento y con la aparición de nuevas terapias como la crioterapia, braquiterapia y termoterapia se espera que las secuelas observadas en esta paciente sean parte de la historia pasada y que se logre disminuir el riesgo de segundas malignidades debido al empleo cada vez menor de radioterapia externa.
PUBERTAD PRECOZ DE ETIOLOGÍA POCO FRECUENTEM.aD. Gómez Bustos, F. Vela Casas y J. Sánchez Calero
Hospital Universitario Virgen Macarena. Sevilla. España.
Antecedente y objetivos: La pubertad precoz es más frecuente en las niñas. En el 90 % de los casos es de origen ideopático; se debe descartar siempre un origen orgánico. Una de las etiologías que hay que excluir sería la neoplásica.
Material y métodos: Niña de 2 años que presenta en los últimos 4 meses aumento del tamaño de las mamas, mayor pigmentación de la areola, desarrollo de los labios mayores, hipertrofia de clítoris y pubarquia en el último mes; distensión abdominal, aumento del apetito, incremento del peso y de la talla, flujo vaginal maloliente acompañado de prurito intenso. En el examen físico: peso: 14 kg (P75-P90). Talla: 93 cm (P75) SC: 0,50 m2. Abdomen: blando, depresible, sin viscreromegalias, discretamente distendido, se palpa masa sólida en hipogastrio. Se aprecia telarquia en el estadio III de Tanner; hipertrofia de clítoris. Exámenes complementarios: alfa-feto proteína y beta-HCG: normal. Marcador tumoral CA19.9: normal. Estudio hormonal: 17 betaestradiol: 681 pg/ml (25–86). FSH: < 0,05 mUI/ml (20–13) LH: < 0,07 UI/ml (1,78-92,10). Testosterona y androstendiona: normal. Radiografía de mano y muñeca izquierdas: edad ósea: compatible con 3 años. Ecografía abdominal y de pelvis: lesión anexial izquierda sólida bien definida con pequeñas áreas quísticas periféricas de 47–50 mm, con vascularización significativa y que desplaza la vejiga urinaria. RM abdominal: masa heterogénea con áreas quísticas y polos sólidos en localización ovárica izquierda. Se encuentra bien delimitada, en contacto con el polo superior del útero. Aumento del tamaño uterino, que muestra una morfología puberal y un engrosamiento del endometrio. Diagnóstico: tumor de ovario izquierdo. Síndrome estrogénico secundario. Tratamiento: extirpación quirúrgica de la masa: salpingooforectomía. Estadiaje: quirúrgico. Se envía muestra para anatomía patológica.
Resultados: Pubertad precoz de origen ovárico; el tumor de la granulosa juvenil es el responsable de la clínica. Se realiza estadiaje quirúrgico encontrándose en estadio la de la clasificación de Figo. Se realiza exéresis de la masa y control: remite la clínica y normalización de los exámenes complementarios.
Conclusiones: El tumor de la gramulosa juvenil es infrecuente en la infancia, representa el 5–8 % de los tumores ováricos a esta edad, suelen ser unilaterales y funcionantes y dan síntomas precozmente, entre ellos la telarquia prematura. Su diagnóstico suele producirse en estadios iniciales, lo que permite el abordaje quirúrgico y condiciona un buen pronóstico. Los parámetros evolutivos más importantes son la clínica y los marcadores sexológicos de actividad ovárica. Estadios avanzados y recidivas ensombrecen el pronóstico.
REVISIÓN DEL RETINOBLASTOMA. RESUMEN DE NUESTRA EXPERIENCIAF. Vela Casas, C. López Calero, A. Campos Barasoaín y J. Sánchez Calero
Hospital Universitario Virgen Macarena. Sevilla. España.
Objetivos: Revisión y análisis retrospectivo de los casos diagnosticados de retinoblastoma (RB).
Material y métodos: Para cada uno de los casos de retinoblastoma se han analizado las siguientes variables: edad en el momento del diagnóstico (dividimos en 3 grupos: menores de 3 meses, entre 3 y 12 meses y mayores de 12 meses), sexo, antecedentes familiares de retinoblastoma, estudio genético, estudio, tamaño del tumor y patrón de crecimiento, síntoma de inicio, unilateral/bilateralidad del tumor, tipo de tratamiento, tipo de esquema de quimioterapia y número de ciclos, toxicidad. Respuesta (dividimos 3 subgrupos: respuesta completa [RC], respuesta parcial [RP], estabilización y progresión), recidivas y supervivencia libre de enfermedad.
Resultados: De un total de 8 casos, 4 se diagnosticaron en menores de 3 meses, 2 entre los 3 y 12 meses y 2 por encima de los 12 meses; 6 eran hembras y 2, varones. Sólo hubo 1 caso con antecedente familiar de RB. Tenían estudio genético realizado 3 pacientes: 2 portadores de una mutación en el gen del RB y otro con cariotipo normal. De los 8 casos, 4 presentaban un estadio E en el momento del diagnóstico y 4, un estadio D. Sólo 1 fue bilateral desde el momento del diagnóstico. En los bilaterales asincrónicos, el período de patencia entre la aparición del primer tumor y el segundo fue variable. En todos los casos, el tamaño del tumor, fue mayor de 1 cm y predominó el patrón de crecimiento endofítico. El síntoma de inicio más frecuente fue la leucocoria. Del total de casos, 4 fueron unilaterales y 4 bilaterales. Se realizó enucleación en todos los unilaterales y en todos los primeros tumores en los casos bilaterales. En los bilaterales, 2 casos de segundos tumores progresaron con crioterapia obteniendo RC con quimioterapia, 1 caso progresó con crioterapia obteniendo RC con enucleación y radioterapia posterior y 1 caso obtuvo RC con radioterapia.
Conclusiones: Encontramos el 50 % de RB bilaterales, cuneado se habla del 25–35 %. La edad media del diagnóstico y la supervivencia en nuestra serie coinciden con lo publicado. Todos los pacientes de nuestra serie sufrieron enucleación al tratarse de estadios avanzados al diagnóstico. Todos estaban libres de enfermedad al final del seguimiento. Con la crioterapia, la quimioterapia y la radioterapia, la mayoría consiguieron conservar el ojo contralateral y la visión, salvo un caso que acabó en doble enucleación.
REVISIÓN DE LA CASUÍSTICA DE TUMORES EN LA ADOLESCENCIA EN UN SERVICIO DE ONCOHEMATOLOGÍA INFANTILN. Naut Suberví, C. Mata Fernández, E. Cela de Julián, C. Beléndez Bieler, A. Cantalejo López y P. Galarón García
Servicio de Oncohematología Infantil. Hospital General Universitario Gregorio Marañón. Madrid. España.
Objetivos: Determinar cuáles han sido las patologías tumorales de la adolescencia tratadas en nuestro hospital, así como su evolución.
Material y método: Se revisan 114 historias de pacientes adolescentes de edades comprendidas entre los 11 y los 17 años, tratados de enfermedades neoplásicas desde 1976 a 2006. Se determina el sexo, la edad, la neoplasia, el tratamiento seguido, la realización o no de trasplante de progenitores hematopoyéticos y la supervivencia libre de enfermedad. Se clasifica la adolescencia como temprana a los 10–13 años; intermedia, a los 14–16 años, y tardía, a los 17–20 años.
Resultados: De los 114 pacientes tratados la edad media de los mismos ha sido de 13 años. El 65 % (74) fueron hombres y el 35 % (40) mujeres. La clasificación por patologías fue: leucemias linfoblásticas agudas: 18 (16 %), linfomas no Hodgkin 12 (10,6 %), osteosarcomas 12 (10,6 %), linfomas de Hodgkin 11 (9,7 %), sarcomas de Ewing 8 (7 %), tumores neuroectodérmicos primitivos 6 (5,3 %), leucemias mieloblásticas agudas 6 (5,3 %), rabdomiosarcomas 5 (4,4 %), linfomas de Burkitt 5 (4,4 %), neuroblastomas 4 (3,5 %), gliomas 5 (4,4 %): 2 cerebrales, 1 del nervio óptico, 2 astrocitomas, tumores de partes blandas no rabdomiosarcomas 5 (4,4 %), sarcoma sinovial 2, indiferenciado 1, mesenquimatosis maligna 1, fibrosarcoma 1, seminoma 2 (1,8 %), histiocitosis de células de Langerhans 2 (1,8 %), carcinoma nasofaríngeo 2 (1,8 %), 1 tumor de Wilms, 1 tumor carcinoide, 1 germinoma pineal, 1 teratoma ovárico benigno, 1 paraganglioma aórtico, 1 carcinoma renal, 1 schwanoma maligno, 1 carcinoma epidermoide, 1 hepatoblastoma, 1 carcinoma medular de tiroides y 1 síndrome mielodisplásico. Todos ellos se trataron en función de los protocolos nacionales-internacionales vigentes en el momento, con quimioterapia, radioterapia y/o cirugía. De los 114 pacientes, 78 se encuentran en remisión completa (69 %) (mayor de 5 años el 64 %, menor de 5 años el 36 %), 26 habían fallecido en el momento del estudio (23 %), 9 se encuentran en tratamiento actual (7,9 %) de los cuales 4 están en tratamiento de recaída y de 1 se perdió su seguimiento.
Conclusiones: En la adolescencia está descrita una mayor incidencia de tumores en varones que en mujeres, en nuestro estudio la relación es de 1,8:1.
Entre los 15 y los 19 años los 3 tumores descritos en la bibliografía como más frecuentes son por orden linfoma de Hodgkin (19 %), leucemia (12 %) y tumores cerebrales y de médula espinal (10 %), En nuestra casuística entre los 15 y 17 años las leucemias han sido las neoplasias más frecuentes seguidas del linfoma de Hodgkin y no Hodgkin. Por debajo de los 14 años la patología más frecuente son las leucemias (linfoblásticas y mieloblásticas). Nuestro estudio corrobora esto, el porcentaje de leucemias es del 23,2 %, seguida de los linfomas (de Hodgkin y no Hodgkin) en el 18,8 %. La supervivencia global de las enfermedades neoplásicas en la adolescencia a los 5 años descrita en la bibliografía es del 73 %. En relación con estos datos, el 69 % de nuestros pacientes se encuentran en remisión completa, el 7,9 % se encuentra en tratamiento actualmente y el 23% ha fallecido.
HEMANGIOENDOTELIOMA HEPÁTICO INFANTILF. Vela Casas, C. López Calero y J. Sánchez Calero
Hospital Universitario Virgen Macarena. Sevilla. España.
Objetivos: Presentar un caso clínico de un tumor hepático muy infrecuente en la infancia.
Material y métodos: Historia clínica y exploración: niña de 11 meses, asintomática que en un control de niños sanos presenta hepatomegalia: lóbulo derecho a 3 cm del reborde costal, en línea media clavicular y lóbulo hepático izquierdo que sobrepasa el ombligo y la línea media. Como diagnóstico diferencial de tumores hepáticos en este grupo de edad pensamos que podría tratarse de: hemangioendotelioma, hamartomas, teratomas (benignos) y hepatoblastoma, tumor rabdoibe, tumor del saco vitelino y rabdomiosarcomas (malignos). Como pruebas complementarias solicitamos: hemograma, tiempos de coagulación y velocidad de sedimentación globular (normales). Bioquímica general: GOT: 41 U/l y FA: 450 U/l. Estudio de lípidos (normal). Alfafetoproteína: 18,8 ng/ml. Beta-HCG: 0,1 mU/ml. Estudio hepático a virus B y C negativo. Radiografía de tórax normal. Ecografía abdominal: masa sólida en lóbulo hepático izquierdo, bien delimitada, de bordes polilobulados de 7,6 cm × 6,5 cm, muy heterogénea y vascularizada. Angiorresonancia nuclear magnética de abdomen con gadolinio: tumor sólido en lóbulo hepático izquierdo, polilobulado de 9 cm × 7 cm × 4,5 cm en sentido crancocaudal, transversal y anteroposterior. Tumor de naturaleza vascular multiseptada con intenso, precoz y permanente realce con gadolinio, inicialmente periférico y progresivamente más central. Antes de estos hallazgos el diagnóstico que se hizo fue de hemangioendotelioma hepático infantil.
Resultados: Debido a que la niña estaba totalmente asintomática la actitud que se tomó fue la observación y el seguimiento estrecho mediante ecografía abdominal y valores de alfafetoproteína y beta-HCG.
Conclusiones: Los tumores hepáticos son muy infrecuentes en la infancia. Dos terceras partes de ellos son malignos. El hemangioendotelioma es el tumor vascular hepático más frecuente en la infancia. Es un tumor muy raro, aunque la mayoría son benignos. Pueden regresar o involucionar. Tienen una elevada mortalidad con relación a su asociación con insuficiencia cardíaca congestiva, el síndrome de Kassabach-Meritt y sangrado intraabdominal. Pueden ser simples o multifocales y de pequeño o gran tamaño. La actitud en las lesiones simples y asintomáticas es la observación y seguimiento. En las lesiones múltiples y sintomáticas: corticoides, interferón alfa segunda quimioterapia, radioterapia, embolización, cirugía y trasplante hepático.
ADOLESCENTES SUPERVIVIENTES DE CÁNCER INFANTIL. EVALUACIÓN PSICOSOCIAL Y SECUELAS A LARGO PLAZOG. Medin, C. Beléndez Bieler, E. Cela de Julián, P. Galarón García, C. Mata Fernández y M.A. Cantalejo López
Servicio de Oncohematología Infantil. Hospital Gregorio Marañón. Madrid. España.
Objetivo: Estudiar las secuelas y las necesidades psicosociales de una población de supervivientes de cáncer pediátrico, así como evaluar su calidad de vida actual. El otro aspecto importante es explorar cuánta información disponen acerca de la enfermedad y el tratamiento por el que han pasado y si creen que esta experiencia ha influido en su vida de alguna forma. Este último aspecto nos parece fundamental a la hora de disponer de información para prevención y conductas saludables a la vez que sabemos que muchos eran niños pequeños en el momento del tratamiento y el tratamiento de la información ha quedado a cargo de los padres, que poseen su propio estilo comunicacional y su propio criterio acerca de que información brindar.
Métodos: Estudio transversal. Recolección de datos: revisión de historias clínicas. Los pacientes se han contactado por teléfono y se ha convocado para revisión y entrevista semiestructurada con el niño y el adulto acompañante. Los adolescentes completaron el cuestionario de calidad de vida CHIP en su versión para adolescentes y el inventario de crecimiento postraumático (PTGI).
Resultados: Población: 40 pacientes con edad promedio en el momento de la entrevista de 17,7 años (12–21), Edad en el momento del diagnóstico 6,3 (0,1-12). Diagnósticos: LLA el 34 %, SNC el 25 %, Wilms el 10 % y otros tumores sólidos el 31 %. Información sobre la enfermedad: el 50 % de los adolescentes habló acerca de su enfermedad, conocía su nombre y usaba la palabra cáncer cuando se refería a su experiencia. Sin embargo, el 23 % de los entrevistados no sabía la enfermedad que habían tenido y el motivo por el que seguían viniendo a los controles, y el 28 % conocía el nombre de la enfermedad pero la familia evitaba intencionalmente el uso de la palabra cáncer y pedía que no se le mencionase al adolescente. Si bien para la mayoría de los padres esta experiencia ha sido crucial y de gran influencia en su vida, sólo el 42 % de los adolescentes manifestó lo mismo y completaron el PTGI. En cuanto a la educación, la mayoría continúa con escolaridad normal (84 %), pero el 15 % perdieron un año y el 13 % 2 años a causa del tratamiento. Un número importante necesita apoyo extraescolar (31 %). El grupo con necesidades escolares especiales corresponde a supervivientes de tumores de sistema nervioso central con secuelas neurocognitivas y/o psiquiátricas. Respecto de la inclusión social, la mayoría están integrados en sus ámbitos, salen con amigos y realizan las mismas actividades que el resto de los adolescentes de su contexto social. Sin embargo, el 41 % son vistos por sus padres o se ven a sí mismos como tímidos, inhibidos y con escasa actividad social. Aproximadamente, un tercio de la población consultó a un especialista de salud mental. Como dato relevante, la mayoría se manifestaba satisfecho con su vida actual, otorgando una puntuación de 7 a 10 cuando fueron preguntados acerca de cómo se sentían con su vida tal cual era.
Conclusiones: Los resultados son alentadores en términos del bienestar de la población. Sin embargo, hay ciertas necesidades relativas a la información, al trabajo con algunas familias y al abordaje terapéutico de algunos adolescentes que se encuentran desatendidas en esta población que se beneficiaría de un programa sistemático de seguimiento de supervivientes.
LINFOMA LINFOBLÁSTICO PRE-B DE PRESENTACIÓN CUTÁNEAM. García de Paso Mora, C. Márquez Vega, G. Ramírez Villar y E. Quiroga Cantero
Unidad de Oncología Infantil. Hospitales Universitarios Virgen del Rocío. Sevilla. España.
Introducción: Los linfomas cutáneos (LC) son aquellos procesos linfoproliferativos malignos –de linfocitos T o B– cuya primera manifestación clínica es la presencia de lesiones cutáneas sin existir enfermedad extracutánea en el momento del diagnóstico y en la que se puede observar afectación ganglionar o visceral en el curso de la enfermedad. Tiene una incidencia de 0,5-1 caso/100.000 habitantes. Hasta el 65 % derivan de células T, sólo el 25 % deriva de células B y el 10 % restante depende de otras células del sistema inmune. La afectación cutánea primaria de LNH fenotipo pre-B es extremadamente infrecuente (< 1 % de todos los LNH), pero aparece con mayor frecuencia en niños que en adultos.
Caso clínico: Presentamos el caso de un niño de 11 años sin antecedentes de interés que consultó por tumoración en cuero cabelludo de 8 meses de evolución sin otra sintomatología acompañante. En la exploración se encontró una masa de partes blandas de 8 × 8 cm en la zona parietooccipital, dura, redondeada, fija y adherida a planos profundos y conglomerado adenopático cervical de 2-3cm. El resto de la exploración fue normal. En pruebas de imagen (ecografía y TC craneal) se aprecia masa que afecta a la piel y al tejido celular subcutáneo sin afectar al cráneo. El estudio de extensión fue negativo, incluyendo TC toracoabdominal, aspirado de médula ósea, citología de LCR y PET. El estudio de laboratorio fue normal. Anatomía patológica (AP): linfoma linfoblástico de células precursoras B (LLCpreB). Inmunohistoquímica: positivo a CD20, TDT, CD34 y vimentina e índice MIB-1 alto. Inicia tratamiento quimioterápico según protocolo EURO-LB02 para estadio II, consiguiendo una remisión completa tras la inducción. Actualmente asintomático y libre de enfermedad al año del diagnóstico. Continúa tratamiento de mantenimiento.
Discusión: El LLCpreB es un subtipo histológico poco frecuente. Afecta fundamentalmente al cuero cabelludo y al cuello. Predomina en mujeres, a diferencia de nuestro paciente, rango de edad 5–15 años, dentro de la cual se encuentra nuestro caso. No afecta a la epidermis, y suele presentarse como enfermedad localizada. Debemos realizar un diagnóstico de extensión y hacer el diagnóstico diferencial fundamentalmente con la leucemia y linfoma de Hodgkin. También es esencial la determinación inmunohistoquímica, ya que es difícil distinguir los diferentes tipos histológicos, incluso a veces diferenciarlos de hiperplasias u otros procesos no oncológicos. El LLpreB expresa CD10, CD19, CD20, CD79a, TDT, CD34, CD43, vimentina y es negativo para marcadores mieloides, de células T y CD30, como ocurría en nuestro paciente. Esta dificultad en el diagnóstico AP ha motivado durante mucho tiempo la existencia de múltiples clasificaciones. En la actualidad, se suele utilizar la unión de dos clasificaciones: la de la Organización Mundial de la Salud y The European Organization for Research and Treatment of Cancer. El tratamiento de elección es la poliquimioterapia. Responden muy bien al tratamiento, con una remisión completa en la mayoría de los casos y un pronóstico excelente.
Comentario: Con este caso pretendemos ilustrar una patología infrecuente pero que plantea dificultades en el diagnóstico diferencial con entidades de mucho peor pronóstico y remarcar la importancia de diagnosticar y seguir las lesiones cutáneas de los niños, ya que constituyen una de las más frecuentes consultas en atención primaria.
ESTUDIO DE MUTACIONES DEL GEN PTCH A PROPÓSITO DE TRES OBSERVACIONESH. Ruiz Larrañaga, B. Calvo Martínez, J. Burgos Bretones, A. Navajas Gutiérrez
Unidad de Investigación y Unidad de Oncología Pediátrica. Hospital de Cruces. Vizcaya. España.
Objetivo: Cada vez son más numerosas las asociaciones de diferentes tumores en un mismo individuo dentro de la edad pediátrica. Aunque la quimioterapia (QT) y la radioterapia (RT) administradas para el primer tumor puedan ser factores precipitantes de nuevas neoplasias, otras veces los predisponentes genéticos subyacen en tumores de novo como sucede en la presentación de características cutáneas de los pacientes presentados. En este estudio tratamos de identificar los hallazgos inmunohistoquímicos y moleculares en la revisión de 3 casos clínicos de 3 varones con neoplasias dermatológicas asociadas a otros tumores.
Material y métodos: Caso 1: varón de 21 años que presentó tumor de fosa posterior cerebral a los 4 años. Se realizó resección completa. La histología confirmó un medulomioblastoma. Recibió QT y RT según protocolo SIOP PNET 2. No presentó evidencia de recidiva tumoral en controles de resonancia magnética (RM) de seguimiento. El 19 de octubre de 1999 ante hallazgo en control de RM de tumoración frontal derecha intraaxial, se realiza resección y se confirma un meningioma fibroso. Ante lesiones sospechosas cutáneas (múltiples nevus) se extirpa el 14 de febrero de 2007 un quiste dérmico en abdomen y, posteriormente, una lesión de partes blandas en región maxilofacial todavía pendiente de clasificar. Caso 2: varón de 14 años diagnosticado de tumor de fosa posterior cerebral el 8 de octubre de 1997. Se realizó exéresis completa del tumor y la histología fue de meduloblastoma clásico. Recibió QT y RT craneoespinal según protocolo SIOP-3 hasta el 22 de diciembre de 1997. No presentó datos de recidiva tumoral en posteriores controles de RM de seguimiento. El paciente y su familia presentan múltiples nevus, por lo que en el seguimiento se observa en exploración física del 29 de junio de 2006 una lesión en el cuero cabelludo de 1 mes de evolución costrosa y con rascado que se decide biopsiar. El diagnóstico histológico es carcinoma basocelular, por lo que se completa cirugía con ampliación de bordes el 9 de febrero de 2007, confirmándose el diagnóstico esta vez con bordes libres de tumor. Se extirpa otra lesión sospechosa en el hombro, con resultado de nevus displásico con márgenes libres. Caso 3: varón de 15 años sin antecedentes de interés que acude al pediatra a los 7 años presentando una lesión cupuliforme en la mejilla izquierda de 2 meses de evolución, indolora. Se refiere al dermatólogo y se extirpa el 22 de septiembre de 1999 con resultado de tumor anexial basalioide. El 3 de abril de 2000 se extirpa otra lesión y, al confirmarse un carcinoma basocelular, se solicita excisión ampliada de la lesión con bordes libres el 9 de enero de 2001. Evoluciona sin complicaciones posteriores. Métodos: se realiza el estudio molecular de gen PTCH e inmunohistoquimica a partir de tejido fijado en bloques de parafina. Se extrae el ADN con un kit comercial. La amplificación completa del gen mediante PCR se realiza con 23 pares de primers (Hahn et al, 1996). Los productos de la amplificación se secuencian en el ABI XL 3130. Se realizan estudios inmunohistoquímicos para la expresión de CD34, PS100, SMA y P53.
Resultados: Pendientes de la finalización del análisis total al escribir este resumen.
Conclusiones:1) Las lesiones dermatológicas malignas en la infancia siguen representando un problema de retraso diagnóstico por parte de familia y pediatras que desconocen su importancia evolutiva. 2) En las lesiones de novo ante la evidencia histológica, el estudio molecular puede facilitar el vigilar aparición de nuevas neoplasias asociadas. 3) En los meduloblastomas con aparición de procesos cutáneos evolutivos, actualmente es posible mediante el estudio molecular seleccionar las poblaciones de riesgo (Gorlin, entre otros).
TUMOR DE WILMS EN UN ADULTO JOVENM. Martínez, Ll. Hernández, F. Almazán y G. Javier
Servicio de Pediatría. Hospital Universitario Germans Trias i Pujol. Badalona. Barcelona. España.
Objetivos: El nefroblastoma o tumor de Wilms es un tumor típico de la infancia. La incidencia anual en adultos se calcula que es menor a 0,2 casos por millón. No hay guías de tratamiento específicas para este grupo de edad, por lo que se suelen seguir protocolos pediátricos.
Material y métodos: Presentamos el caso clínico de un paciente de 21 años afectado de tumor de Wilms y remitido desde el servicio de urología para valoración y tratamiento.
Resultados: Varón de 21 años que consultó por abdominalgia de 10 meses de evolución. En la ecografía abdominal y RM se detectó masa renal izquierda con trombo que invadía la vena renal y vena cava inferior hasta la aurícula derecha. A las 2 semanas presentó insuficiencia cardíaca derecha junto con tromboembolismo pulmonar que requirió intervención quirúrgica urgente (nefrectomía radical y extracción de trombo pulmonar). Pese a la sospecha diagnóstica de hipernefroma, el estudio anatomopatológico fue diagnóstico de tumor de Wilms sin anaplasia con infiltración de cápsula renal, vena renal izquierda y vena cava inferior. En el estudio de extensión se detectaron tres metástasis hepáticas y una adenopatía paraórtica. Se inició tratamiento quimioterápico según protocolo SIOP-2001 de tumor de Wilms estadio IV. Al mes persistía una de las metástasis hepáticas que se extirpó quirúrgicamente. Durante el tratamiento presentó como principales complicaciones un episodio de tiflitis y una pequeña hemorragia subaracnoidea a consecuencia de plaquetopenia posquimioterapia. Al cabo de 6 meses de haber finalizado el tratamiento (cirugía + quimioterapia + radioterapia) y al año y medio de la nefrectomía, permanece en remisión completa.
Conclusiones: Pese a que el tumor de Wilms en adultos tiene una frecuencia de presentación extremadamente baja, es un diagnóstico que debe ser tenido en cuenta, especialmente en adolescentes o adultos jóvenes afectados de una masa renal. De esta forma, se conseguiría llegar al diagnóstico de forma más precoz y evitar el diagnóstico de la enfermedad en estadios avanzados como suele ocurrir en estos pacientes.
PROTOCOLO DE TRATAMIENTO DE LOS GLIOMAS DE MAL PRONÓSTICO CON IRINOTECÁN EN COMBINACIÓN CON CISPLATINOJ. Mora 1, O. Cruz1, A. Parareda1, C. De Torres1, A. Guillén2, R. Navarro2, G. García2 y J.M. Costa2
1Servicio de Oncología y 2Neurocirugía. Hospital Universitari Sant Joan de Déu. Universidad de Barcelona. Barcelona. España.
Objetivos: Los astrocitomas son un grupo de neoplasias para las cuales el tratamiento adyuvante es aún controvertido. Después de un ensayo piloto con pacientes diagnosticados de astrocitoma medular en progresión, en el que se observó que la combinación de irinotecán y cisplatino podía ser efectiva (Mora et al, Neuro Oncol. 2007), iniciamos en 2004 un ensayo de fase II para todos los pacientes con astrocitoma de alto riesgo. El objetivo primario es determinar la eficacia antitumoral y el perfil de seguridad de I/C en pacientes con astrocitoma.
Pacientes y métodos: Se incluyen pacientes con diagnóstico de astrocitoma de alto grado al inicio o en recaída, astrocitoma de bajo grado en progresión sintomática y tumores intrínsecos de tronco. El tratamiento consiste en la administración semanal en régimen ambulatorio de C (30 mg/m2) seguida de irinotecán (65 mg/m2), hasta un total de 16 dosis. Los pacientes con astrocitoma de alto grado y tumor de tronco al debut reciben el tratamiento estándar con radioterapia y temozolamida intercalado tras las primeras 8 dosis.
Resultados: Han sido incluidos 17 pacientes, 5 con astrocitoma grado I de la OMS, 6 con astrocitoma grado II, 3 con astrocitoma grado III y 3 que no fueron biopsiados (2 intrínsecos de tronco y 1 de vías ópticas). Ninguno de los pacientes presentaba signos o historia de neurofibromatosis. Las localizaciones primarias: 4 mesencafálicos, 3 de tronco, 3 de vías ópticas, 1 cerebelar y 6 espinales. Se realizó una resección macroscópicamente completa en 1 caso, subtotal en 4 y en 9 sólo biopsia. Los efectos secundarios fueron vómitos fácilmente controlados salvo en los pacientes con tumores de tronco. Sólo 2 pacientes no han completado el protocolo por alergia al cisplatino. En 11 pacientes (65 %) se obtuvo una respuesta clínica completa de los síntomas neurológicos incluyendo un tumor de tronco y todos los medulares. En 12 (70 %) se observó una reducción de las medidas del tumor al final del tratamiento incluyendo 2 pacientes con astrocitoma de bajo grado y de línea media con una disminución del volumen > 50 % y cambios en la imagen, lo que sugería remielinización. Un caso de tronco progresó durante el tratamiento. Ninguno de los pacientes con astrocitoma de bajo grado ha precisado radioterapia y todos están vivos y con mejoría clínica, media de seguimiento 1 año.
Conclusiones: Con el protocolo irinotecán/cisplatino se han conseguido respuestas clínicorradiológicas remarcables y evitar la radioterapia en los pacientes con astrocitoma de bajo grado.
POLIPOSIS FAMILIAR (PAF) Y RIESGO DE CÁNCER DE COLON EN LA INFANCIA Y ADOLESCENCIA: ¿CUÁNDO REALIZAR LA COLECTOMÍA PROFILÁCTICA?J. Balaguer1,2, S. Oltra3, A. Segura4, M. García4, A. Pereda5 y V. Castel1
1Unidad de Oncología Pediátrica, 2Fundación para la Investigación. 3Unidad Genética. 4Unidad del Consejo Genético Cáncer. 5Unidad de Gastroenterología Pediátrica. Hospital Universitario La Fe. Valencia. España.
Introducción: La PAF es una enfermedad hereditaria autosómica dominante con una elevada penetrancia y diferente variabilidad clínica. Existe una mutación en el exón 15 del gen APC,y dependiendo de la localización de la mutación se divide en FAP atenuada o clásica (dentro de la clásica existe un grupo de mayor riesgo asociado a FAP grave). Se caracteriza por pólipos colorrectales que pueden malignizarse (hasta el 15 % en la adolescencia). Es necesario un seguimiento mediante colonoscopia a partir de los 13 años o en caso de clínica. Requieren colectomía profiláctica en algunos casos, pero existe discusión acerca del momento óptimo, puesto que dará lugar a un síndrome de intestino corto. Por otro lado, no debe demorarse más de lo necesario por la probabilidad de degeneración maligna.
Resultados: Existen 26 familias PAF (con 56 portadores) en el registro de la Unidad de Consejo Genético del Cáncer (UCGC) de nuestro hospital. De ellas, 4 tienen 5 portadores diagnosticados durante la infancia o adolescencia; 3 pacientes están asintomáticos y 2 son portadores de una misma familia presentaron rectorragia al diagnóstico. Una paciente de 8 años que fue remitida por dolor abdominal periumbilical de 2,5 años de evolución y rectorragia. Sin antecedentes de interés salvo que su padre tenía poliposis intestinal. En la exploración clínica destaca dolor a la palpación profunda en FII, tacto rectal limpio. En el enema opaco se aprecia una poliposis difusa colónica y en el tránsito intestinal completo se ven, además, pólipos en íleon terminal. Se realiza colonoscopia y se biopsian 2 pólipos sésiles que se informan como adenomatosos. En el fondo de ojo se aprecian manchas leopardo temporales a las máculas de ambos ojos (hipertrofia pigmentaria de retina). Se realiza test genético en la familia. Resultan portadores la paciente, su padre y su hermano de la mutación c. 3927_3931 del AAAGA (codón 1309), relacionada con la FAP grave. Continúa siendo controlada estrictamente mediante colonoscopia y se aprecia un aumento del número de pólipos de recto-sigma 2 años después, por lo que se inició tratamiento con sulindac a 100 mg/d, con una reducción transitoria. En el último control, la paciente tiene 21 años, en la colonoscopia se aprecian miles de pólipos frágiles que llegan hasta la línea pectínea, algunos de más de 15 mm. Dada la persistencia ocasional de las deposiciones con sangre, y el aumento de pólipos se ha recomendado una colectomía total con anastomosis ileoanal. Actualmente, el hermano de 10 años, también portador, está siendo controlado por rectorragia, estando pendiente de colonoscopia diagnóstica (en el enema opaco se visualizan pólipos). El padre de los pacientes desarrolló un adneocarcinoma de colon, por el que fue intervenido a los 45 años.
Conclusiones:1) El estudio de mutaciones del gen APC debe considerarse una prueba diagnóstica obligatoria en los niños pertenecientes a familias de riesgo. 2) Los niños portadores deben ser controlados estrictamente y a partir de los 12 años o ante aparición de clínica debe realizarse una colonoscopia diagnóstica. 3) En caso de poliposis clínica sintomática, se debe valorar el momento óptimo de la colectomía profiláctica para que interfiera lo menos posible en el desarrollo psicomotor y prepuberal del paciente, pero sin diferirla más de lo conveniente.
TRATAMIENTO DEL SARCOMA DE EWING CON EL PROTOCOLO MSKCC P6 MODIFICADOJ. Mora1, O. Cruz1, A. Parareda1, C. De Torres1, R. Huguet2, A. Montaner3, A. Guillén4 y R. Navarro4
Servicios de 1Oncología, 2Ortopedia, 3Cirugía y 4Neurocirugía. Hospital Universitario Sant Joan de Déu. Universidad de Barcelona. Barcelona. España.
Objetivos: Los tumores de la familia del sarcoma de Ewing responden al tratamiento intensivo con agentes alquilantes. El protocolo MSKCC P6 está basado en ciclofosfamida y adriamicina alternado con ifosfamida y etopósido, además de radioterapia a todos los pacientes. La supervivencia a 5 años publicada con este protocolo es del 82 % en pacientes con enfermedad no metastásica. Presentamos nuestra experiencia con una versión modificada.
Pacientes y métodos: El protocolo original se modificó en que sólo se administran 2 ciclos (en lugar de 3) de ifosfamida y etopósido para disminuir el riesgo de tumores secundarios; las dosis de ifosfamida son más elevadas (2,8 g/m2/d × 5 d en lugar de 1,8) en los casos de pobre respuesta histológica(< 90% necrosis), y la adriamicina se administra previa infusión del cardioprotector dexrazoxane. Además, según las alteraciones moleculares de cada tumor se añaden agentes biológicos: rapamicina, imatinib y bifosfonatos.
Resultados: Desde diciembre de 2001 hasta enero de 2007 se han incluido 19 pacientes, 4 en recaída. Todos los tumores expresan genes de fusión con EWS. En el momento de la inclusión 3 eran metastáticos (2 en médula ósea y 1 en el pulmón bilateral). De los 16 no metastáticos, 1 era primario de tentorio, 6 de esqueleto axial y 9 locorregionales (> 200 cc) no axiales. La media de edad a la inclusión en el protocolo es de 10 años. La respuesta radiológica después del tercer ciclo fue de remisión completa o parcial (> 50 %) en 15 de 19 (79 %); respuesta menor (< 25 %) en 1 y 3 no eran evaluables. La resección quirúrgica se realizó en 15 (79 %) casos. La respuesta histológica posquimioterapia fue evaluable en 10 y en todos excepto 1 caso fue superior al 90 % de necrosis. Todos los pacientes han recibido radioterapia y 1 consolidación con altas dosis y rescate autólogo. De los 19 pacientes, 3 han fallecido y 16 están vivos (4 en tratamiento). Un paciente (estadio 4) progresó durante el tratamiento y 5 pacientes (26 %) han recaído, media 20 meses, 4 sistémicos y 1 local (segunda). Todos los pacientes con recaída entraron en remisión con una tercera línea de tratamiento y 2 están libres de enfermedad. Para los 15 pacientes tratados al diagnóstico (2 metastáticos), la supervivencia libre de eventos (SLE) es del 80 % y la supervivencia global del 93 %, media seguimiento de 19 meses desde la inclusión. De los 4 pacientes incluidos en recaída, 2 fallecieron, 1 vive con la enfermedad y 1 está libre de ella. La SLE y supervivencia global del grupo entero es del 68 y 85 %, respectivamente, media de seguimiento 22 meses. No se ha documentado toxicidad irreversible ni segundos tumores.
Conclusiones: Con este protocolo se han conseguido supervivencias superiores a las reportadas en sarcoma de Ewing evitando la toxicidad asociada a protocolos de tratamiento incluyendo terapia con altas dosis y trasplante y manteniendo la posibilidad de rescatar las recaídas con nuevos tratamientos citotóxicos.
SARCOMA SINOVIAL: UN TUMOR SÓLIDO DE PREADOLESCENTESL. Moreno-Martín Retortillo, MaM. Andrés Moreno, A. Cañete Nieto y V. Castel Sánchez
Unidad de Oncología Pediátrica. Hospital Infantil La Fe. Valencia. España.
Introducción: El sarcoma sinovial (SS) es una entidad infrecuente en la población pediátrica que predomina en adolescentes a diferencia de otros sarcomas de partes blandas (SPB). La escasez de casos, la dificultad de cirugía exitosa y la dudosa quimiosensibilidad hacen difícil su tratamiento y oscurecen su pronóstico, especialmente en casos de enfermedad metastásica. Presentamos nuestra experiencia con SS en los últimos 20 años.
Casos clínicos: Describimos 4 casos de niños de 7, 9, 11 y 13 años de edad afectados de SS de las extremidades con cirugías exitosas (primarias o secundarias) que recibieron distintos regímenes de quimioterapia. El primer caso se inició con 9 años (tras 3 años de síntomas), consiguió resección completa tras dos cirugías y recayó 5 años después (local y pulmonar). El segundo caso falleció por progresión de enfermedad metastásica. Los dos casos siguientes (13 y 7 años) consiguieron cirugía exitosa y el último recibió quimioterapia adyuvante. Ambos se encuentran libres de enfermedad actualmente.
Discusión: El SS es una entidad que afecta típicamente a adolescentes y preadolescentes a diferencia de otros SPB. Habitualmente, su diagnóstico es dificultoso, pues los síntomas osteomusculares son atribuidos a patología traumática/deportiva. Estos pacientes suelen ser valorados por distintos especialistas hasta que se llega al diagnóstico. Es preciso que la cirugía se realice en centros de referencia para conseguir la resección completa. El tratamiento adyuvante es controvertido, pero necesario en los casos con afectación regional o metastásica, en los que la remisión es difícil de conseguir. Además, su seguimiento se debe prolongar más de lo habitual, pues son frecuentes recaídas tardías (caso 1).
CARCINOMA NASOFARÍNGEO EN EL ADOLESCENTEC. Sábado1, M. Arguis2, J.L. Dapena1, M. Delgado1, S. Gallego1, L. Gros1, M. Ramos2, J. Giralt2 y J. Sánchez de Toledo1
1Servicio de Oncología-Hematología Pediátrica. 2Servicio de Oncología Radioterápica. Hospital Vall d'Hebron. Barcelona. España.
Introducción: La oncología de la adolescencia, además de tratar entidades patológicas distintas de las de los niños más pequeños, comporta unos problemas sociales y personales propios de los adolescentes.
Objetivos: Presentamos una serie de 5 pacientes adolescentes afectos de carcinoma nasofaríngeo tratados en nuestro centro en los últimos 10 años que ilustran las peculiaridades de la oncología del adolescente.
Métodos: Se identificaron en la base de datos del servicio los pacientes afectados de carcinoma nasofaríngeo. Se revisaron las historias clínicas. Todos los pacientes recibieron tratamiento con cisplatino, 5 fluoruracilo, ácido folínico y radioterapia.
Resultados: En todos los casos, la clasificación histológica del carcinoma nasofaríngeo fue del tipo indiferenciado. La edad al diagnóstico fue de 17 años en 1 caso y 15 años en los 4 restantes. La forma de presentación en todos los casos fue tumoración y dolor de 5 meses de evolución como media (rango 3–7 meses). Del total, 3 casos correspondían a estadio III y otros 2 a estadio II y IV, respectivamente. Todos los pacientes presentaron mucositis grado IV y requirieron alimentación parenteral y analgesia con opiáceos. Dos pacientes requirieron cuidados intensivos: 1 por un cuadro de insuficiencia respiratoria al inicio y otro por sepsis y neumonía causada por Capnocytophaga spp. y S. aureus. Hubo 2 pacientes que recibieron amifostina como radioprotector y otro precisó una dosis de rasburicasa por hiperuricemia. Hay 1 paciente que tiene hipotiroidismo subclínico como efecto secundario y todos ellos presentan xerostomía. Una paciente presentó descompensación de un trastorno psiquiátrico durante el tratamiento y su transición a la edad adulta ha sido muy inadecuada. Todos siguen en remisión completa con un seguimiento medio de 8 años y 4 meses (4 años 3 meses- 10 años 9 meses).
Conclusiones: El carcinoma nasofaríngeo es un tumor poco frecuente pero no excepcional en los adolescentes. Suele presentarse en estadios avanzados tras una historia prologada de síntomas y signos. A pesar de ello, el pronóstico en niños y adolescentes es bueno. La forma histológica indiferenciada es casi constante. La toxicidad de los regímenes utilizados es muy alta y merece estudios colaborativos para disminuir las graves toxicidades agudas y las secuelas a largo plazo.
TRATAMIENTO CONSERVADOR DEL TUMOR DE SENO ENDODÉRMICO VAGINAL EN LA INFANCIAM. Villa Alcázar1, M. Pacheco Cumani1, J.C. Ollero Fresno2, J.A. García Asensio2 y B. López-Ibor Aliño1
1Unidad de Oncología Pediátrica. Hospital Montepríncipe. 2Hospital San Rafael. Madrid. España.
Objetivos: Ratificar que la biopsia inicial y el tratamiento con bleomicina, etopósido y carboplatino puede inducir una respuesta tumoral completa (incluyendo normalización de la αFP) en pacientes con tumores germinales malignos de vagina, y evitar la cirugía, que, aun siendo conservadora (vaginectomía parcial), puede condicionar la función reproductora y/o sexual de las pacientes.
Caso clínico: Niña de 10 meses de edad que acude al hospital por un cuadro de sangrado vaginal intermitente de 15 días de evolución tras haber ido en varias ocasiones a otros servicios de urgencias, donde se había llegado al diagnóstico de posibles abusos sexuales. La paciente es remitida por su pediatra al servicio de cirugía de nuestro hospital, donde se solicita una ecografía y RM pélvica en la que se objetiva una masa de 57 × 31 × 41 mm en vagina. Se procede a la biopsia y resección subtotal de la tumoración. El diagnóstico de anatomía patológica es: tumor del seno endodérmico. El estudio de extensión es negativo y la cifra de αFP es de 63.293 UI/ml. La paciente recibe 6 ciclos de quimioterapia consistente en VP-16 120 mg/m2/día días + 1 a + 3, carboplatino 600 mg/m2/día el día + 2 y bleomicina 15 mg/m2/día el día + 3. Tras el tercer ciclo se normaliza la αFP y tras el cuarto ciclo se encuentra en remisión por RM. Al finalizar el tratamiento de quimioterapia se realiza vaginoscopia, en la que no se aprecian restos tumorales. La paciente sigue reevaluaciones periódicas con RM/ecografías y determinación de αFP. Se encuentra en remisión completa 3 años y medio después del fin de tratamiento.
Resultados: La paciente sigue reevaluaciones periódicas con ecografías/RM y determinación de αFP. Se encuentra en remisión completa 3 años y medio después del fin de tratamiento.
Conclusiones: Los tumores germinales malignos (TGM) en los niños representan el 3 % de los tumores malignos pediátricos. Sólo el 3–8 % de estos tumores son vaginales. La mayoría aparecen en niñas menores de 3 años y se suelen manifestar como sangrado vaginal. La histología es, en la mayoría de los casos, tumor del seno endodérmico. La αFP es un buen marcador para evaluar la respuesta al tratamiento y la situación de remisión completa. Aunque algunos autores mantienen que la cirugía conservadora seguida de quimioterapia es la mejor opción para estos pacientes, nosotros, como otros autores, pensamos que retrasar la cirugía hasta después de un tratamiento de quimioterapia basada en platino es el tratamiento de elección. Con dicho tratamiento, hasta el 35 % de los pacientes pueden alcanzar la remisión completa y evitarla cirugía, manteniendo intactas sus funciones sexuales y reproductivas.
¿ES DISTINTO EL CÁNCER EN NIÑOS MAYORES DE 10 AÑOS?L. Gros, S. Gallego, M. Delgado, C. Sábado, J.L. Dapena y J. Sánchez de Toledo Codina
Servicio de Oncología-Hematología Pediátrica. Hospital Vall d'Hebron. Barcelona. España.
Objetivos: Estudiar la incidencia de los distintos grupos de tumores en pacientes con edad en el momento del diagnóstico mayor o menor de 10 años. Valorar si la edad en el diagnóstico (< o > de 10 años) influye de forma significativa en la supervivencia de forma global y en cada uno de los grupos de tumores.
Material y métodos: Estudio retrospectivo sobre los datos recogidos en nuestra base de datos de niños diagnosticados y tratados en nuestro servicio de tumores sólidos malignos durante los últimos 10 años (1996–2006). Se establecen 2 grupos de pacientes según la edad en el momento del diagnóstico según sea menor o mayor de 10 años. Se compara el número de casos para cada grupo, la distribución por sexos en cada grupo y el porcentaje de supervivientes global para cada grupo de edad. Se compara el valor porcentual de cada grupo de tumores para cada grupo de edad y el porcentaje de supervivientes de cada grupo de tumores en cada uno de los dos grupos de edad.
Resultados: Se analizan los datos de 705 pacientes: 463 < 10 años (65 %) y 242 > 10 años (35 % del total: el 30 % entre 10 y 16 años y el 5 % entre 16 y 20 años). En el grupo de < 10 años el 54,6 % son niños y el 45,3 % niñas; en los > 10 años, el 52 % son varones, y el 48 %, mujeres. El porcentaje de supervivientes es del 80 % en los < 10 años y del 83% en los > 10 años. En ambos grupos el grupo de tumores del SNC es el más numeroso, con un valor similar (27 frente a 29).En el grupo de < de 10 años son importantes los neuroblastomas (17 %), los retinoblastomas (10 %), sarcomas de partes blandas (9,5 %) y los LNH (8,2 %). En los > de 10 años los 3 grupos con mayores porcentajes son los linfomas Hodgkin (21 %), los LNH (13,6 %) y los tumores óseos (11,2 % de forma global). En cuanto a la supervivencia, hemos apreciado diferencias en los porcentajes de supervivencia en el grupo de los tumores del SNC con peor supervivencia en los pacientes menores (71,9 frente a 81,5), en el grupo de tumores óseos con peores supervivencias para los mayores (osteosarcoma: 66 frente a 46/síndrome de Ewing: 84,6 frente a 62,5). En otros grupos con un número de pacientes en cada uno suficientemente representativo (LNH, neuroblastoma, TMM), no hemos hallado diferencias en el porcentaje de supervivientes.
Conclusiones: Nuestros pacientes han sido tratados de forma homogénea. Las enfermedades son las mismas en general en niños de menos o más de 10 años, si bien existen, como es conocido, tumores más frecuentes en cada grupo de edad: para pacientes menores (neuroblastoma, retinoblastoma, tumores renales) y tumores más presentes en la segunda década de la vida (tumores óseos, enfermedad de Hodgkin y carcinomas). En la supervivencia, sólo en los tumores de SNC los menores presentan peor supervivencia, probablemente con relación a tipos histológicos más indiferenciados. En los mayores sólo se aprecia una peor supervivencia en los tumores óseos, tal vez por factores biológicos o por retraso en el diagnóstico.
CARCINOMATOSIS MENÍNGEA EN LA EVOLUCIÓN DE UN NEUROBLASTOMA NEONATAL ESTADIO 4M. Pacheco Cumani1, M. Villa Alcázar1, N. Sanz Villa2, J. Navalón Burgos3 y B. López-Ibor Aliño1
1Unidad de Oncología y Hematología Pediátricas. Hospital Madrid Montepríncipe. 2Servicio de Cirugía Infantil y 3Servicio de Radiología. Hospital San Rafael. Madrid. España.
Objetivo: Presentación de un caso de evolución favorable de una carcinomatosis meníngea en la evolución de un neuroblastoma multicéntrico neonatal estadio 4 (INSS).
Caso clínico: Niño de 15 meses de edad, diagnosticado a los 4 días de vida de neuroblastoma multicéntrico estadio 4 (INSS) n-myc (−). Estudio de extensión al diagnostico: masa suprarrenal izquierda de 1,5 × 1,1 × 1,8. Masa paraespinal derecha 3,8 × 4,9 × 4 cm que se introduce por agujeros de conjunción. Múltiples metástasis hepáticas. Tratamiento recibido: protocolo EINS-99. 1 ciclo de QT (VP-16/CBDCA) a los 12 días de vida por Ph score > 1 (taquipnea y edemas). Respuesta al tratamiento: muy buena respuesta parcial disminuyendo las lesiones hepáticas, desaparición de las masas SR y paraverteberal. Mejoría clínica. Evolución: tras una progresión inicial hepática a los 3 meses de vida (no se administra QT por score Ph 0), disminución progresiva de la hepatomegalia y de las cifras de catecolaminas y enolasa hasta agosto de 2005, en que progresa. Presentación clínica de la progresión: crisis de llanto y decaimiento progresivo hasta coma grado I-II. Localización recidiva: SNC (carcinomatosis meníngea, nódulo en la unión bulbomedular con hidrocefalia (se coloca una DVP). Paravertebral L1 a L3 hepática. Tratamiento de la recidiva: protocolo EINS-NB93. 1) Quimioterapia: VP-carboplatino × 4 ciclos, CFM-ADR × 4 ciclos. Respuesta al tratamiento: muy buena remisión parcial. 2) Cirugía suprarrenal izquierda: el 24 de marzo de 2006 (neuroblastoma con maduración; N-myc neg). 3) Quimioterapia posoperatoria: VP-carboplatino × 1 ciclo (26 de abril de 2006); CFM-ADR × 1 ciclo (22 de mayo de 2006). Fin de tratamiento de quimioterapia: 26 de mayo de 2006. 4) Radioterapia (tomoterapia): en región paraverterbral derecha y en unión bulbomedular ha recibido 1.500 cGy (150 cGy/día). Duración del tratamiento: del 10 de julio de 2006 al 21 de julio de 2006. 5) Altas dosis de vitamina A: inicio el 15 de septiembre de 2006 × 2 ciclos. Suspendido en diciembre de 2006 por HIC benigna. Situación actual: remisión completa.
Conclusiones: El riesgo de recidiva en el SNC del neuroblastoma es muy bajo (< 8 %). A los 3 años del diagnóstico, suele producirse en el contexto de diseminación tumoral en la fase terminal de la enfermedad, en cuyo caso es fatal. La afectación neuromeníngea en un neuroblastoma neonatal es excepcional, así como la evolución favorable del paciente que describimos.
EXPERIENCIA Y CONSECUENCIAS DEL CÁNCER INFANTIL EN LOS JÓVENES SUPERVIVIENTESJ.A. Salinas, M. Guibelalde, M. García e I. Hernández
Hemato-Oncología Pediátrica. Hospital Universitario Son Dureta. Palma de Mallorca. España.
Objetivo: Descripción de los hábitos de salud, comportamientos y consecuencias psicosociales en una cohorte de adolescentes supervivientes de cáncer infantil
Pacientes y método: Encuesta a los 38 participantes, mayores de 13 años, supervivientes diagnosticados de un tipo de cáncer cuando eran menores de 13 años (leucemias, linfomas, tumores sólidos y del SNC) en la unidad de oncología pediátrica, ocurrido entre 1987 y 2004, actualmente en remisión continuada de más de 2 años. Los criterios adicionales del estudio han sido: el que fuesen accesibles telefónicamente, el consentimiento a su participación y que no estuviesen actualmente en tratamiento.
Variables del estudio: edad, sexo, tiempo transcurrido desde el diagnóstico, tipo de cáncer, estudios y trabajo alcanzados, encuesta sobre hábitos de salud (dieta, ejercicio, tabaco, alcohol) y valoración psicoafectiva y social y de la percepción corporal.
Resultados: Características: 25 varones (65 %), 13 mujeres (35 %). Edad (años) en la entrevista: 6 entre 13 y 15 años(16 %), 20 entre 16 y 19 años (52 %), 12 > 20 años (32 %). Diagnósticos: 15 leucemias y linfomas (39 %), 11 tumores SNC (29 %), 11 con otros tumores sólidos (29 %). En el 23 %, el diagnóstico se efectuó cuando tenían menos de 5 años de edad y en el 63 % fue hace más de 10 años. Se determinó el grado de estudios (en el 48 % sólo el grado obligatorio) y la situación laboral (sin trabajo en el 58 %). Los adolescentes presentan conductas de riesgo en cuanto a falta de actividad física (50 %), consumo de tabaco (13 %) y drogas (2, que equivalen al 5 %) y una alimentación inadecuada (15 %), pero niegan el consumo habitual de alcohol (98 %).
Se han identificado categorías de experiencias negativas como: 1) Dificultades con los amigos, familia y estudios. 2) Sentimientos desagradables en relación con la apariencia física. Se han identificado el 37 % de categorías de experiencias positivas como: El tipo de vínculo afectivo positivo, especialmente con la madre, autoestima, sensibilidad a los problemas de los demás y madurez. La adaptación social: el modo en que el joven se relaciona con los padres es bueno en la mayoría, pero en menor proporción con los amigos, y la mayoría se sienten sobreprotegidos.
Conclusiones:1) La percepción de sí mismos ha sido predominantemente positiva. 2) Nuestros resultados sugieren que los adolescentes supervivientes de cáncer en la edad pediátrica necesitan intervenciones de salud dirigidas a fomentar hábitos saludables como son los relacionados con el tabaco, el alcohol, el ejercicio y la dieta. 3) Algunos adolescentes que han padecido cáncer (13 %) además aumentan sus riesgos de secuelas o segundos tumores con hábitos de riesgo como el tabaco. 4) Los adolescentes experimentan no sólo experiencias negativas, sino positivas como consecuencia del cáncer padecido en la edad pediátrica. 5) Encontramos que no siempre la gravedad de las secuelas físicas se corresponden con la alteración en el comportamiento psicosocial. 6) Se enfatiza la necesidad de refuerzo psicológico a los adolescentes que han padecido cáncer y que presenten dificultades de integración social y desarrollo personal, debido a las secuelas dejadas por la enfermedad y los tratamientos.
SUPERVIVENCIA Y SECUELAS DEL CÁNCER INFANTIL. NUESTRA CASUÍSTICAM. Leyva Carmona, M.A. Vázquez López, F. Lendínez Molinos, M. Rodríguez, P. Aguilera, P. Oliva y A. Bonillo Perales
Hospital Torrecárdenas. Almería. España.
Introducción: La supervivencia del cáncer infantil (CI) ha aumentado en los últimos años, hasta estimarse superior al 70 %. La alta tasa de curación trae consigo el aumento de patología secundaria a la enfermedad o a la terapia recibida, que, en ocasiones no se objetiva hasta años después de finalizado el tratamiento. Aunque hay publicadas numerosas revisiones con relación al estado de los supervivientes del CI en relación a diversos aspectos médicos, neuropsicológicos, legales y sociales, la verdadera incidencia de los efectos tardíos no se conoce y, en general, está infraestimada por la pérdida del seguimiento de los pacientes.
Objetivos: Describir la evolución de los niños diagnosticados de cáncer en nuestro hospital y analizar las secuelas en los supervivientes.
Material y métodos: En el período comprendido entre enero de 1972 y junio de 2006 se han registrado en nuestro centro 296 niños (175 varones y 121 mujeres) entre 0 y 14 años con diagnóstico de cáncer. Desconocemos la situación actual de 22 (7,43 % pérdidas) y en los 274 restantes hemos recogido datos con relación a la edad en el momento del diagnóstico, el sexo, el tipo de neoplasia, el tratamiento recibido, la aparición de recidiva, fallecimiento y causa del mismo. Analizamos la supervivencia global y aparición de secuelas, tipo y gravedad de las mismas. El período de seguimiento medio ha sido de 5,62 + /– 4,83 años.
Resultados: La edad media en el momento del diagnóstico fue de 5,44 + /– 3,99 años. La distribución según tipos de neoplasias fue: leucemias (37,2 %), tumores del SNC (14,9 %), neuroblastomas (9,1 %) renales (4,4 %), sarcomas (6,1 %), óseos (5,1 %), retinoblastomas (1,7 %), hepáticos (0,7 %), histiocitosis y germinales (2 %), respectivamente. Recibieron quimioterapia el 74,3 %, radioterapia el 30,4 %, cirugía el 44,3 % y trasplante de médula ósea el 12,3 %. Recidivaron 58 casos (21,17 %). Fallecieron 69 casos (25,2 %), producidos por causa infecciosa (38,8 %), hemorrágica (10,1 %), enfermedad progresiva (33,3 %), y neurológica (17,8 %). La supervivencia global obtenida a los 7 años del diagnóstico fue del 74,9 %. Han presentado secuelas 79 pacientes (38,5 %), de las cuales 40 (19,5 %) fueron consideradas graves. De éstas, las más frecuentes fueron las neurológicas observadas en 9 niños (22,5 %), seguidas por las endocrinas 7 niños (17,5 %), sensoriales 5 casos (12,5 %), segunda neoplasia 8 (20 %), renales 4 (10 %), hepáticas 4 (10 %), músculo-esqueléticas 2 (5 %) y psicológicas 1 (2,5 %). Un total de 9 niños presentaron más de una secuela (11,4 %).
Conclusiones:1) El cáncer pediátrico se cura en una alta proporción. 2) El elevado porcentaje de secuelas graves es preocupante no sólo por la incapacidad y necesidad de atención médica continuada que precise, sino también por la repercusión en la proyección profesional y personal de los enfermos. 3) Debe promoverse la creación de equipos multidisciplinarios y la vigilancia continua de los supervivientes del CI para detectar precozmente los problemas crónicos derivados de su enfermedad y tratamiento, y elaborar programas que faciliten la inserción social de estos de estos pacientes.
EFICACIA Y SEGURIDAD DEL TRATAMIENTO PROLONGADO CON TEMOZOLOMIDA EN PACIENTE CON NEUROFIBROMATOSIS TIPO I AFECTADA DE GLIOBLASTOMA MULTIFORME CEREBRALN. García de Andoin Barandiaran, J.J. Úriz Monaut, N. Arostegi Kareaga, I. Miner Kanflanka y R. Guerrero Pereda
Unidad de Oncología Infantil. Servicio de Pediatría. Hospital Donostia. San Sebastián. España.
Introducción: La temozolomida es un agente metilante de ADN con buena actividad antitumoral, con capacidad para atravesar la barrera hematoencefálica y estable a pH gástrico ácido, lo que permite una biodisponibilidad oral del 100 %. Es conocido el uso de temozolomida como coadyuvante de radioterapia y de forma paliativa en el tratamiento de los gliomas tanto en adultos como en niños y en estos últimos los resultados son bastante pobres. Recientes estudios realizados en adultos han demostrado la seguridad del tratamiento con temozolomida de forma prolongado durante más de 1 año, con mínimos efectos secundarios. Exponemos el caso de una niña afectada de glioblastoma multiforme recidivante tras extirpación completa y radioterapia, con restos tumorales tras cirugía que consigue remisión de la enfermedad después de iniciar tratamiento con temozolomida.
Caso clínico: Niña de 7 años afectada de neurofibromatosis tipo I, con antecedentes de hamartoma de hemisferio cerebeloso derecho y glioma del nervio óptico izquierdo. Presenta cuadro de cefalea y vómitos matutinos de 15 días de evolución, y es diagnosticada de proceso expansivo en el lóbulo frontal derecho en RM. Se realiza exéresis completa del tumor con resultado anatomopatológico (AP) de xantoastrocitoma con imágenes de anaplasia, grado II (necrosis, índice bajo mitótico y Ki 67 > de 1). RM de control poscirugía: extirpación del tumor con captación de gadolíneo lineal en el margen quirúrgico de probable etiología posquirúrgica. A los 3 meses comienza con clínica de cefalea y paresia facial. En RM proceso expansivo con componentes sólidos y quísticos de 5,4 × 4,8 × 4,5 cm con edema perilesional. Se realiza exéresis macroscópica total con resultado AP de transformación a glioblastoma multiforme (con aumento de número de mitosis atípicas y desaparición de la trama de reticulina e índice de ki 67 elevado). RM poscirugía: cambios posquirúrgicos con existencia de focos de hemorragia subaguda y captación lineal de contraste que pueden considerarse hallazgos normales en estudio posquirúrgico precoz. Recibe radioterapia con dosis total 60 Gy durante 47 días junto con temozolomida oral a 75 mg/m2/día. En RM de control tras radioterapia se aprecia recidiva tumoral que es intervenida con exéresis parcial (AP: glioblastoma multiforme). Se decide iniciar tratamiento paliativo con temozolomida oral a 200 mg/m2/día durante 5 días, con ciclos cada 28 días. A partir del segundo ciclo precisa disminuir la dosis a 160 mg/m2 por plaquetopenia. En una RM de control a los 6 meses, no hay evidencia de recidiva o resto tumoral. En la actualidad la paciente ha completado 1 año de tratamiento sin signos de recidiva, con buena tolerancia a la temozolomida y sin efectos secundarios.
Comentarios:1) La temozolomida es un fármaco que puede ser bien tolerado y seguro en el tratamiento de tumores infantiles. 2) A pesar de la indicación de la temozolomida en nuestra paciente como tratamiento paliativo, no sólo se ha conseguido frenar el crecimiento de los restos tumorales, sino que se ha logrado la remisión.
CARCINOMA DE TIROIDES. EXPERIENCIA DEL HOSPITAL SANT JOAN DE DÉU DE BARCELONAA. Laguna1, O. Cruz1, J. Mora1, A. Parareda1, C. de Torres1, S. Segura1, R. Díaz2, A. Montaner3 y M. Vancells3
Servicios de 1Oncología, 2Endocrinología y 3Cirugía. Hospital Universitario Sant Joan de Déu. Universidad de Barcelona. Barcelona. España.
Objetivos: Comunicar la experiencia adquirida en carcinoma de tiroides en la población pediátrica y adolescente en los últimos 10 años.
Introducción: Las neoplasias de tiroides son poco frecuentes en pediatría, aunque tienen mayor incidencia en los adolescentes. El tratamiento multidisciplinar de estos pacientes requiere de protocolos para su diagnóstico, tratamiento y seguimiento, y existen pocas referencias en la edad pediátrica.
Pacientes y métodos: Se realiza un estudio retrospectivo de patología tumoral tiroidea diagnosticada en nuestro centro en pacientes entre 0 y 18 años, en los últimos 10 años.
Resultados: Desde 1997 hasta febrero de 2007 se han diagnosticado 18 neoplasias de tiroides. Adicionalmente, se controla 1 paciente de 2 años con mutación RET y tiroidectomía profiláctica, sin carcinoma hasta el momento. La población de afectados fue de predominio femenino en el 81 % (mujeres n = 13), con edades que oscilaron entre 1 y 16 años, con una media de 12. Se detectó una incidencia superior de tumores diferenciados de tiroides, 16 carcinomas diferenciados (CD) (papilar, el 47 %, y folicular, el 29 %), y 2 carcinomas medulares de tiroides, portadores de mutación RET. Los pacientes con carcinoma medular presentaron una menor edad al diagnóstico (2 y 12 años). El síntoma más frecuente fue el hallazgo de nódulo tiroideo o bocio. La localización más frecuente fue el lóbulo derecho. Todos los CD cursaron con tiroglobulinas elevadas y los medulares con elevación de calcitonina. En la mayoría de los casos se realizó inicialmente una hemitiroidectomía, pero finalmente en el 70 % se practicó tiroidectomía total. En 2 casos se extirparon restos de tiroides guiado por sonda radiactiva. En 7 casos de CD se efectuó tratamiento ablativo con radioyodo (43 %). El 50 % de los CD presentaban metástasis en el momento del diagnóstico, en su mayoría adenopatías locorregionales y en 1 caso pulmonares (6 %). La complicación principal es el hipotiroidismo, ya que el 100 % de los casos requirió tratamiento hormonal sustitutivo. La supervivencia es del 100 %, tras un seguimiento medio de 4 años.
Conclusiones: El CD continúa siendo la causa más frecuente de tumores de tiroides en la edad pediátrica, con predominio en los adolescentes. El tratamiento actual logra una adecuada respuesta. Hemos desarrollado un protocolo para estandarizar el diagnóstico, tratamiento y seguimiento de estos pacientes.
MASA EN OVARIO CONTRALATERAL TRAS COFORECTOMÍA POR CARCINOMA EMBRIONARIOX. Duarte X, G. Caldas, A. Teixeira y M. Chagas
Instituto Portugués de Oncología de Lisboa. Portugal.
Objetivo: Recordar la importancia de establecer un diagnóstico con seguridad antes de adoptar cualquier decisión terapéutica.
Caso clínico: Presentamos el caso de una adolescente de 13 años, remitida en diciembre de 1998 desde Cabo Verde para estudio por una tumoración abdominal de 6 meses de evolución. Los estudios de imagen realizados evidenciaron una voluminosa masa pélvica, que causaba una ureterohidronefrosis bilateral, más acusada en el lado izquierdo. Los marcadores tumorales estudiados (6-fetoproteína, 8-HCG, CEA y CA 125) fueron negativos. El estudio por microscopia electrónica del material obtenido por citología aspirativa con aguja fina estableció como diagnóstico más probable el de carcinoma embrionario de ovario. En el estudio de extensión realizado se excluyó la existencia de metástasis a distancia. Tras un ciclo de quimioterapia con bleomicina, etopósido y carboplatino (BEP), con evidente reducción tumoral, se decidió realizar oóforo-salpinguectomía izquierda. En el acto quirúrgico se constató importante infiltración tumoral de la pared posterior del útero y de la pared anterior del recto, consiguiendo sólo su resección parcial. El estudio anatomopatológico informó de un carcinoma embrionario de ovario, con necrosis de cerca del 90 % del tumor. Finalizó la quimioterapia en abril de 1999, tras cuatro ciclos BEP. El estudio por TC realizado en junio de 1999 reveló una rarefacción de la pared de la ampolla rectal, y microcalcificaciones y atrofia del riñón izquierdo. En el seguimiento posterior hasta julio de 2003 se objetivó regresión de las lesiones de la pared del recto antes descritas.
En enero de 2004 presentó un episodio súbito de dolor abdominal, por lo que se realizó ecografía y TC abdominal, que revelaron una masa sólida heterogénea en el ovario derecho, con implantes peritoneales a lo largo de la sutura de laparotomía. El marcador tumoral CA 125 estaba elevado, con el resto de marcadores negativos. Se propone el diagnóstico de recidiva tumoral tardía con diseminación peritoneal.
Resultados y conclusiones: La paciente es operada en enero de 2004. Se discute el diagnóstico y el abordaje terapéutico.
ESTUDIO BUCODENTAL DE NIÑOS Y ADOLESCENTES EN TRATAMIENTO ONCOLÓGICOM. Franquet1, A. Parareda2, O. Cruz, J. Mora2, C. de Torres2, S. Segura2, A. Laguna2 y A. Cahuana1.
Servicios de 1Odontopediatría y 2Oncología. Hospital Universitari Sant Joan de Déu. Universidad de Barcelona.
Objetivos: El estado de salud bucodental en un paciente oncológico puede alterarse seriamente durante el proceso de la enfermedad, debido a la misma, a la terapia antineoplásica o al abandono de medidas higiénicas. La asistencia odontológica protocolizada disminuye de manera significativa las complicaciones orales agudas y crónicas de estos niños y adolescentes, mejorando sus perspectivas de salud bucodental. En este estudio analizamos las complicaciones bucodentales de los pacientes con cáncer tratados en nuestra institución, en las diferentes etapas de la enfermedad.
Pacientes y métodos: Todos los pacientes diagnosticados de cáncer del desarrollo durante 2006 han sido evaluados de su estado bucodental, incluyendo: 1) Al inicio: educación, motivación y prevención de la higiene oral; valoración de la patología oral preexistente y necesidad de tratamiento ambulatorio u hospitalario con anestesia general. 2) Durante la terapia oncológica: tratamiento de las urgencias dentales de forma conservadora; prevención y tratamiento de las complicaciones orales agudas. 3) Postratamiento oncológico: tratamiento dental conservador y evaluación de las secuelas.
Resultados: Se han incluido un total de 147 pacientes. El porcentaje de casos de niños oncológicos respecto al total de las consultas odontológicas del hospital ha sido del 8,45 %. La distribución de los casos por edad muestra un rango de 5 meses a 20 años. El porcentaje de pacientes con patología oral preexistente al inicio, una mala higiene dental en la mayoría de casos, fue superior al 90 %. Durante la terapia neoplásica, el 82 % de pacientes desarrollaron al menos una nueva lesión oral, principalmente mucositis, xerostomía o queilitis. Durante el año 2006, a diferencia de los anteriores, no se presentó ningún caso de mucositis que requiriera nutrición parenteral.
Conclusiones: La atención odontológica del paciente oncológico al inicio y a lo largo del proceso de la enfermedad tiene un impacto significativo en la salud oral y minimiza los efectos secundarios del tratamiento. Nuestra experiencia nos ha permitido elaborar una pauta de asistencia estructurada para estos pacientes.
UN CASO POCO FRECUENTE DE TUMOR DE CÉLULAS GERMINALESX. Duarte, A. Teixeira, G. Caldas, A. Lacerda, A. Neto, F. Pereira, M.J. Ribeiro y M. Chagas
Instituto Portugués de Oncología de Lisboa. Portugal.
Objetivo: Destacar la importancia del estudio citogenético en los tumores de células germinativas y el problema que nos planteó en el caso que presentamos.
Caso clínico: Adolescente de 14 años y sexo femenino que presentaba un voluminoso tumor pélvico, que podía palparse a nivel del mesogastrio e hipogastrio. Tras las exploraciones complementarias realizadas se estableció el diagnóstico de tumor de células germinativas del ovario derecho. En el estudio de extensión se descartó la existencia de metástasis a distancia.
Se realizó oóforo-salpinguectomía derecha tras quimioterapia neoadyuvante, con resección completa del tumor. El estudio citogenético de la pieza operatoria informó un cariotipo 46,XY. Se realizó un cariotipo constitucional, que también fue 46,XY. En los estudios genéticos posteriormente realizados se evidenció la existencia de una microdelección en Yp11.23, con la supresión de la secuencia codificante del gen determinante del sexo (SRY).
Resultados y conclusiones: La disgenesia gonadal completa (síndrome de Swyer) es una situación genética poco frecuente que se caracteriza por presentar un cariotipo 46,XY y fenotipo femenino, sin alteraciones somáticas, y asociada a un elevado riesgo de neoplasias gonadales. La paciente fue sometida a una segunda intervención quirúrgica para la exéresis profiláctica de la gónada izquierda.
CARCINOMA EPIDERMOIDE EN UN PACIENTE CON EPIDERMÓLISIS AMPOLLOSAF. Vela Casas, C. López Calero y J. Sánchez Calero
Centro de Trabajo: Hospital Universitario Virgen Macarena. Sevilla.
Objetivos: Presentar un caso clínico de carninoma epidermoide en un adolescente con epidermólisis ampollosa distrófica recesiva.
Material y métodos: Adolescente de 16 años con epidermólisis ampollosa distrófica recesiva que presenta una lesión ulcerada en el dorso de la mano derecha de bordes costrosos y elevados con fondo ulcerado y sucio de evolución tórpida hace 1 año. Presenta tumoración axilar derecha, con eritema, distensión de la piel y aumento temperatura local desde hace 1 mes.
Antecedentes personales: Epidermólisis ampollosa distrófica recesiva. Estenosis esofágica. Anemia ferropénica crónica. Desnutrición calórica. Epistaxis ocasionales. Exploración: palidez de piel y mucosas. Piel con descamación y ulceraciones generalizadas debidas a su patología de base. En el dorso de la mano derecha presenta una úlcera de 4 cm de diámetro de bordes costrosos, centro ulcerado y con secreción purulenta. Linfedema del miembro superior derecho. En la axila derecha se palpa una tumoración que abarca la axila y la pared costal derecha, dura y con la piel a tensión y enrojecida. Atrofia muscular marcada en extremidades y anquilosis en articulaciones metacarpofalángicas e interfalángicas proximal y distal de ambas manos. El resto de la exploración por aparatos, sin hallazgos. Diagnóstico diferencial: abceso secundario a úlcera de evolución tórpida o metástasis de carcinoma epidermoide. Inicia tratamiento antibiótico con amoxicilina-ácido clavulánico durante 2 semanas y posteriormente con cloxacilina oral sin mejoría. La tumoración crece después de 20 días de tratamiento. Juicio clínico: carcinoma epidermoide con metástasis ganglionar axilar. Pruebas complementarias: Hemograma: Hb 9,7g/dl; Hlc: 29,9%; VCM: 85,90; leucocitos: 12.400/mm3; neutrófilos: 8.900/mm3; plaquetas: 482.000/mm3. Bioquímica: Calcio: 12,5 mg/dl. PT: 8,4 g/dl. PCR: 53 mg/l. El resto es normal. Estudio de coagulación: normal. Ecografía axilar: flemón inflamatorio en axila derecha de 7,5 cm. Radiografía de tórax: aumento de partes blandas en la región axilar derecha que parece comprimir los arcos costales derechos. Mínimo pinzamiento del seno costodiafragmático derecho. TC de tórax y axila derecha. Derrame pleural derecho de escasa cuantía. Masa axilar derecha de 8,5 cm de eje mayor, con necrosis interna y captación heterogénea de la parte sólida. No se aprecia solución de continuidad con la musculatura intercostal derecha (entre los arcos costales 2° y 3°) produciendo un discreto desplazamiento de la pleura en esa zona. La musculatura de la pared costal derecha se encuentra desestructurada, por lo que habría que descartar infiltración tumoral o proceso inflamatorio. En la axila izquierda también se aprecian algunas adenopatías aumentadas de tamaño biopsia de la lesión ulcerada en el dorso de la mano derecha: carcinoma poco diferenciado. La positividad por IHQ para CK, aunque focal confirma la naturaleza epitelial del tumor. Citología por punción aspiración con aguja fina de la tumoración en axila derecha: hallazgos citológicos indicativos de carcinoma de células escamosas. Diagnóstico definitivo: carcinoma espinocelular estadio III.
Resultados: Se interviene al paciente resecando el tumor primario y la metástasis ganglionar axilar.
Conclusiones: Ante una úlcera de evolución tórpida y con características de malignidad en un paciente con epidermólisis ampollosa, hay que pensar en el Carcinoma espinocecular como primer diagnóstico, ya que las genodermatosis son un factor de riesgo para este tipo de tumores.
UNA UNIDAD DE ONCOLOGÍA PEDIÁTRICA EN LA SANIDAD PRIVADA: CIENCIA Y HUMANISMO POR Y PARA EL NIÑO DIAGNOSTICADO DE CÁNCERM. Villa Alcázar, M. Pacheco Cumani, C. Pitillas Salvá, C. Bengoechea Menéndez, P. Montes Muñoz y B. López-Ibor Aliño
Unidad de Hematología y Oncología Pediátrica. Hospital Madrid Montepríncipe. Madrid. España.
Una unidad de oncología pediátrica no es una sección o servicio pediátrico propiamente dicho. No es un lugar, no es un departamento dentro de un hospital. No. Es un equipo multidisciplinar que trabaja para que la atención al niño y adolescente diagnosticados de cáncer y a su familia sea integral. Trabaja con el objetivo de que el niño tratado de un cáncer llegue a ser un adulto sano tanto desde el punto de vista físico, como psíquico, social y espiritual. Un adulto sano con los mismos derechos y deberes que sus coetáneos que no estuvieron enfermos. El cáncer infantil no sólo afecta al niño, sino que afecta a toda su familia y entorno social. Por eso, debemos atender también a sus necesidades y extender nuestro trabajo al domicilio y al colegio.
Todo comenzó en el año 1992, hace ya 15 años. Iniciamos una unidad de oncología pediátrica en un pequeño hospital de Madrid llamado La Zarzuela, integrada en el servicio de pediatría. Solo atendíamos a los enfermos que pertenecían a Sanitas, una aseguradora medica.
En 1999 la unidad se trasladó al Hospital San Rafael, donde creció al atender a niños de todos los seguros médicos privados. En el año 2006, la unidad se trasladó al Hospital Madrid Montepríncipe dando un paso adelante significativo en cuanto a su infraestructura técnica, científica y humana. Los requisitos para que una unidad de oncología pediátrica pueda funcionar adecuadamente están publicados por la SIOP y nuestra unidad los cumple sobradamente. En el momento actual el equipo está integrado por tres médicos oncólogos pediatras, un psicólogo y un musicoterapeuta, todos con dedicación exclusiva, enfermería entrenada en oncología pediátrica, UVI pediátrica y un voluntariado perteneciente a la AECC. La Unidad está integrada en un servicio de pediatría con todas las especialidades pediátricas. Existe una UCI oncológica dentro de la unidad, dos habitaciones para TMO autólogo y una sala de procedimientos/sedaciones. El grupo de padres Mil y una esperanzas vela por la atención de sus hijos en nuestro centro. La unidad de oncología funciona integrada en el Centro Integral de Cáncer Clara Campal (CIOC), en el que se desarrollan proyectos de investigación de genética molecular del cáncer y desarrollo de nuevas drogas. La filosofía de trabajo consiste en “adaptar el hospital al niño, no el niño al hospital”. Por ello, la estancia media es de 3,5 días, el hospital de día funciona de 9 a 21 horas los 7 días de la semana y las consultas de 9 a 6 los días laborables. Contamos con una unidad de cuidados paliativos domiciliaria y atendemos a los padres en un grupo específico si el niño muere. Los oncólogos pediatras están localizados las 24 horas del día. Consiste además en “adaptar la medicina al enfermo” es decir, practicar una oncología personalizada en la que el tumor recibe el tratamiento que necesita y en la que el enfermo recibe la atención y cuidados que tanto él como su familia requieren para integrar la enfermedad en la vida normal del niño y su familia. Todos los enfermos, sin excepción, están registrados en el RNTI y registrados y tratados con protocolos nacionales o internacionales según su patología.
Se trata de una unidad de oncología pediátrica en el contexto exclusivo de la sanidad privada española, que por sus características funcionales permite adaptarse a las necesidades del niño y su familia. Presentamos los datos de incidencia, tratamiento y supervivencia que, asimismo, figuran en el RNTI.
PANENTEROCOLITIS POR IRINOTECÁN EN PACIENTE AFECTADO DE HEPATOPATÍA CRÓNICA Y POLIMORFISMO UGT1A1 TA7/TA6E. López Rodrigo, R. Castro Medina, C. de Torres, S. Segura, O. Cruz y J. Mora
Servicio de Oncología. Hospital Universitario Sant Joan de Déu. Universidad de Barcelona. Barcelona. España.
Objetivo: Ilustrar, a propósito de un caso, un cuadro de toxicidad grave por irinotecán en un paciente tratado por recaída de rabdomiosarcoma alveolar de paladar y con hepatitis crónica. Irinotecán es un profármaco que se metaboliza en SN-38, convirtiéndose en un potente inhibidor de la topoisomerasa I. SN-38 se metaboliza mediante glucuronoconjugación en el hígado y se elimina por vía biliar, conversión catalizada en gran medida por la enzima UGT1A1. En estudios farmacogenéticos se ha descrito que la inserción de dos nucleótidos (TA) extra en la región TATA del promotor de UGT1A1 en heterocigosis (TA6/TA7) y sobre todo en homocigosis (TA7/TA7), causa del síndrome de Gilbert, se relacionan con mayor toxicidad por irinotecán. La función hepática alterada también se ha asociado a mayor toxicidad por irinotecán.
Material y métodos: Se ha analizado la longitud del repeat TA de la región promotora mediante PCR. Asimismo, se han analizado SNP (single nucleotide polipmorphisms) tanto en el promotor como en el resto del gen UGT1A1. Se está analizando en la actualidad UGT1A2 y UGT1A9, así como la enzima CYP3A4.
Resultados: Presentamos el caso de un paciente de 12 años diagnosticado de recidiva local de rabdomiosarcoma alveolar en paladar tras 6 años de remisión y hepatitis crónica secundaria al tratamiento quimioterápico previo. Tras resección quirúrgica se inicia tratamiento con irinotecán y carboplatino. Inicia clínica gastrointestinal con vómitos, diarreas y dolor abdominal que requiere ingreso al octavo día de inicio del tratamiento. Evoluciona a íleo paralítico, fallo hepático con edema generalizado y ascitis. Se asocia neutropenia febril con anemia y trombopenia. El tratamiento incluye ileostomía de descarga con resección ileal parcial por necrosis isquémica. Se objetivan cambios macroscópicos y microscópicos compatibles con panenterocolitis tóxica y cirrosis hepática. El estudio funcional hepático con DISIDA demostró un enlentecimiento en la curva de eliminación biliar. El análisis farmacogenético mostró el polimorfismo de UGT1A1 TA7/TA6. No se han hallado SNP ni en el promotor ni en la región codificante de UGT1A1. Con el tratamiento de soporte después de 7 meses se ha conseguido la recuperación de la integridad intestinal tanto anatómica como funcional.
Conclusiones: El tratamiento con irinotecán requiere la detección y tratamiento precoz de la clínica gastrointestinal. La detección de factores de riesgo incluyendo marcadores genéticos, estudios de imagen funcional hepática y fármacos coadyuvantes debe realizarse con el fin de ajustar la dosificación. El uso de antibióticos orales de amplio espectro puede reducir la actividad betaglucuronidasa intestinal y, con ello, el riesgo de toxicidad intestinal.
