
Barcelona, viernes 5 de marzo y sábado 6 de marzo de 2004
Consumo de alcohol durante el embarazo en España: análisis de los potenciales riesgos para defectos congénitos de diferentes dosis de alcohol ingerido durante el embarazo
M.L. Martínez-Frías1,2, J.L. Frías3, E. Rodríguez-Pinilla1 y E. Bermejo1
1Centro de Investigación sobre Anomalías Congénitas (CIAC). Instituto de Salud Carlos III. 2Facultad de Medicina de la Universidad Complutense de Madrid. España. 3Department of Pediatrics, University of South Florida, Tampa, Estados Unidos.
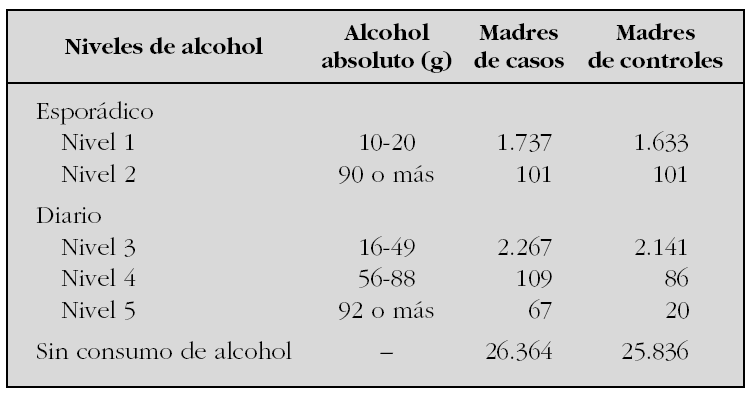
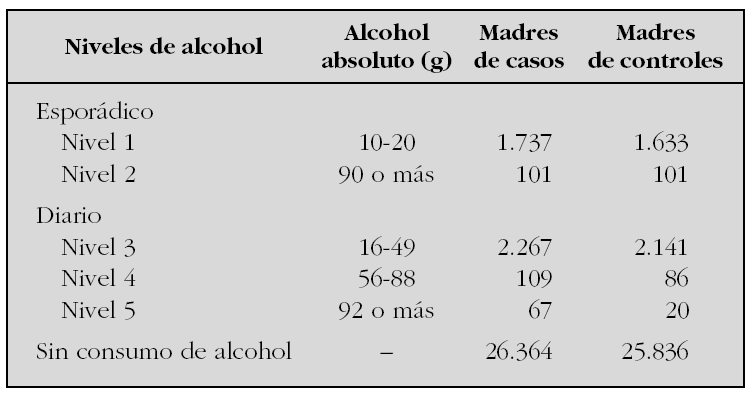
Utilizando los datos del Estudio Colaborativo Español de Malformaciones Congénitas (ECEMC), analizamos la evolución del consumo de alcohol a lo largo de 27 años en España. Para ello, se han establecido cinco grupos de madres en función de la periodicidad y las cantidades ingeridas durante el embarazo. Las cantidades se decidieron siguiendo las normas que establecen que una unidad de bebida estándar de vino/cerveza tiene 10 g de alcohol absoluto y una de bebida fuerte (ginebra, etc.), 20 g de alcohol absoluto, y teniendo en cuenta las cantidades consumidas por cada madre. Los cinco grupos fueron: dos con cantidades crecientes de exposiciones esporádicas y tres con cantidades crecientes de dosis diarias. En la tabla se exponen los grupos con sus dosis y las muestras de cada grupo y tipo de madres que las consumieron.
La distribución temporal del consumo durante el embarazo muestra una tendencia decreciente y estadísticamente muy significativa de la proporción de madres que beben de cada grupo, excepto en el grupo 5. Esta tendencia es similar en todas las comunidades autónomas. Por otra parte, se observa que en cada nivel de alcohol la proporción de madres que beben es significativamente diferente entre las distintas comunidades. Es decir, que no se bebe igual ni en las mismas cantidades en todas las autonomías. Analizamos el potencial riesgo que podrían tener cada una de las cantidades de alcohol establecidas para cada uno de los 12 defectos congénitos seleccionados. Los resultados muestran que, incluso dosis bajas y esporádicas, podrían suponer un riesgo para algunos defectos. En todas las dosis estudiadas se observaron riesgos para diferentes defectos congénitos que se aumentan en la medida que se incrementan las dosis. Este trabajo representa el primer estudio epidemiológico que demuestra que dosis muy bajas de alcohol podrían alterar el desarrollo embrionario-fetal. Y aunque es posible que sea en función de la fracción de la población genéticamente susceptible, como no podemos identificarla, el mensaje que se debe transmitir es que cero alcohol es la única dosis segura durante el embarazo.
Bibliografía
Martínez-Frías ML, et al. Evolución temporal y por comunidades autónomas del consumo de diferentes cantidades de alcohol durante el embarazo. Med Clin (Barc). 120:535-41.
Martínez-Frías ML, et al. Risk for congenital anomalies associated with different sporadic and daily doses of alcohol consumption during pregnancy. En prensa.
Exposición materna a misoprostol y secuencia de Möebius en el recién nacido: ¿un problema emergente en españa?
A. Pérez Aytes1, P.D. Iglesias1, F. Moreno2 y J.L. Carbonell3
1Hospital Infantil La Fe. 2Mediterránea Médica. Valencia. 3Centro Municipal de Bienestar Social. Torrejón de Ardoz, Madrid. España.
Introducción: Diversos trabajos realizados en varios centros de Brasil han confirmado una fuerte relación entre exposición materna a misoprostol y síndrome de Möebius en el recién nacido. Se presenta un caso de síndrome de Möebius tras la ingestión materna de misoprostol, que pensamos que representa la "punta del iceberg" de un problema que, en nuestra opinión, no ha sido suficientemente valorado en nuestro país fuera de ciertos ámbitos profesionales.
Caso clínico: J.G. es un varón nacido de una tercera gestación en una madre de 20 años sana. Durante el embarazo la madre intentó abortar, tomando, por vía oral, un total de 24 comprimidos de misoprostol (1 comprimido = 200 μg). Los comprimidos los tomó a lo largo de 2-3 días, hacia la 5.ª-6.ª semana de gestación. Tras la ingesta presentó fuertes dolores abdominales durante al menos una semana. No presentó metrorragias. El intento de aborto fracasó y el embarazo continuó su curso sin otros problemas especiales. No existía adicción a drogas. Parto hospitalario, espontáneo, de presentación cefálica. Nació con un peso de 3.570 g, test de Apgar 9/10. Desde el principio llamaba la atención que no parpadeaba y tenía una cara "inexpresiva", por lo que se pensó que podía tener algún tipo de déficit visual. A los 3 meses fue diagnosticado de síndrome de Möebius. RM cerebral: normal. Cariotipo: 46;XY, normal. A los 8 meses presentaba buen estado de nutrición, era un niño activo, mantenía la cabeza y se mantenía sentado. En la exploración neurológica se observaban signos de parálisis bilateral característicos de los nervios craneales VII (facial) y VI (oculomotor externo o abducens): facies inexpresiva, con ausencia de mímica facial al llanto o ante cualquier estímulo (VII par craneal), estrabismo convergente bilateral y ausencia de "mirada lateral" (VI par craneal). El tono muscular y los reflejos osteotendinosos eran normales, no había defectos de reducción en extremidades y no se apreciaban otras anomalías.
Comentario: El misoprostol es un análogo de la prostaglandina E1, que bajo el nombre comercial de Cytotec®, se utiliza ampliamente en medicina como protector de la mucosa gástrica. Además de esta acción, el misoprostol induce contracciones en el útero gestante y actúa también dilatando el cuello uterino, todo lo cual ha hecho que, bajo control médico, se utilice para expulsión de restos fetales en caso de aborto diferido y para la interrupción voluntaria de embarazo. A partir de 1990 se popularizó el uso del misoprostol como sustancia abortiva clandestina, fuera del control médico, en Brasil y otros países sudamericanos. Su efecto teratógeno está ampliamente reconocido y los patrones malformativos que se asocian con más frecuencia son: síndrome de Möebius, pies zambos, artrogriposis, defectos de reducción distales en extremidades, y sindactilia. El uso de misoprostol como abortivo clandestino, como ocurrió en el caso aquí presentado, se ha introducido en España y nos consta que se está usando ampliamente. Las especiales circunstancias que suelen rodear estos casos llevan a ocultar inicialmente su uso a los profesionales sanitarios, cosa que también ocurrió en esta ocasión, y esto hace difícil la auténtica valoración del problema.
Ácido valproico y gestación: problemas en la identificación de teratógenos
E. Rodríguez-Pinilla, C. Mejías, G. Dequino, P. Fernández, B. Rato y M.L. Martínez-Frías
Centro de Investigación sobre Anomalías Congénitas (CIAC). Sección de Teratología Clínica y Servicios de Información sobre Teratógenos. Instituto de Salud Carlos III. Madrid. España.
Es bien conocido que el tratamiento con anticonvulsivantes en embarazadas epilépticas conlleva un incremento del riesgo no sólo para malformaciones congénitas (mayores y menores), sino también para el retraso mental. La mayoría de los fármacos anticonvulsivantes comercializados presentan un efecto teratogénico (trimetadiona, fenitoína, ácido valproico, carbamazepina). La identificación y cuantificación del riesgo teratogénico en el ser humano de los distintos anticonvulsivantes se ha venido realizando gracias a los sistemas de vigilancia epidemiológica y estudios epidemiológicos sobre recién nacidos (vivos y/o muertos) con defectos congénitos. Con los avances del diagnóstico prenatal y la posibilidad de interrumpir legalmente la gestación en caso de malformaciones fetales, los sistemas de registro de malformaciones en recién nacidos (vivos o muertos) y estudios caso-control están perdiendo la posibilidad de identificar y, en su caso, cuantificar, el efecto teratogénico de los medicamentos que se vayan comercializando. En la Base de Datos del Estudio Colaborativo Español de Malformaciones Congénitas (ECEMC) hemos analizado, en dos períodos de tiempo, el riesgo para malformaciones tras la exposición materna a anticonvulsivantes, teratógeno reconocido, durante el primer trimestre de la gestación. Nuestros resultados muestran una reducción de los riesgos en el segundo período, consecuencia del diagnóstico prenatal seguido de la posibilidad de interrupción de los embarazos de fetos afectados. Los riesgos obtenidos para cualquier defecto congénito tras la exposición materna a anticonvulsivantes en los dos períodos de tiempo han sido en el primer período: OR: 7,4 (3,5-15,7); p < 0,00001; segundo período: OR: 2,5 (1,3-4,8); p = 0,0023. Los riesgos para espina bífida en concreto, igualmente en ambos períodos, han sido en el primer período: OR: 9,89 (1,5-65,7); p = 0,0065; y en el segundo período: OR: 2,5 (0,2-28,2); p = 0,23. Por otra parte, disponer de una serie consecutiva de recién nacidos malformados tiene una gran ventaja en relación a las series de casos establecidas sobre pacientes de centros pediátricos, quirúrgicos, de genética, etc. Esa ventaja se debe a que en las series consecutivas se dispone del espectro completo de malformaciones de los niños que presentan los diferentes síndromes o los diferentes efectos de las exposiciones maternas. Se presentan las manifestaciones clínicas de todos los recién nacidos malformados de la serie consecutiva del ECEMC (80 casos con malformaciones) y se discuten los problemas de los estudios sobre teratógenos al no estudiarse las interrupciones de las gestaciones de fetos afectados, junto con las implicaciones que ello conlleva en el uso racional de fármacos durante la gestación y en el asesoramiento genético.
Presentación de 2 nuevos casos con síndrome de Saavedra
M.D. Ontiveros y M. Orera Clemente
Unidad de Genética. Hospital General Universitario Gregorio Marañón, Madrid. España.
Introducción: En 1992 Saavedra et al describieron un nuevo síndrome teratogénico producido por la exposición prenatal a éteres de glicol, específicamente al metilcellosolve y al etilenglicol (MC y EG), en 44 jóvenes mexicanos hijos de mujeres obreras, que trabajaron con estos agentes químicos durante la gestación. El estudio epidemiológico mostró que los solventes MC y EG cumplían todos los requisitos para ser considerados teratógenos. En el estudio clínico multidisciplinar se describieron los principales hallazgos del síndrome (facies peculiar, alteraciones esqueléticas, retraso psicomotor). En el estudio experimental se corroboró su capacidad teratogénica y la relación dosis-efecto y se correlacionaron las alteraciones producidas en las dos especies: seres humanos y roedores.
Objetivo: Presentar 2 casos nuevos, de origen europeo, con este síndrome teratogénico.
Casos clínicos:Caso 1. Niña de 13 años de edad que desde el nacimiento presentó fenotipo peculiar y retraso psicomotor, no clasificados previamente. En la exploración física se observó una paciente cuyas características físicas, psicológicas y cognitivas similares a las descritas en los pacientes mexicanos con exposición prenatal a éteres de glicol. Llama la atención que en este caso, quien estuvo expuesto a los solventes fue el padre pero, como fue comprobado posteriormente, estos se impregnaban en la ropa y eran absorbidos, vía cutánea por la madre. También en el líquido prostático se acumulaban estos químicos.
Caso 2. Niño de 10 años de edad, con retraso psicomotor grave y alteraciones fenotípicas que afectaban al sistema musculoesquelético y al sistema nervioso central, similares a las observadas en los casos graves del síndrome de Saavedra. En este caso la madre estuvo en contacto durante todo el embarazo a dos altas de MC y EG unido a otros solventes orgánicos y agentes tóxicos.
Discusión: El conocimiento de este síndrome debe alertar por el uso cada vez mayor e indiscriminado de agentes químicos, como los éteres de glicol, e insistir en el uso de medidas laborales preventivas, tanto en varones como en mujeres, para evitar la aparición de nuevos casos.
¿Dónde se esconde el síndrome de Kabuki?
I. Lorda-Sánchez, C. García-Arévalo, V. Martínez-González, M. Rodríguez de Alba, C. Ramos Corrales y C. Ayuso
Servicio de Genética. Fundación Jiménez Díaz. Madrid. España.
El síndrome de Kabuki (KS; OMIM 147920) fue descrito por primera vez en 1981. Su diagnóstico se basa en cinco criterios principales: 1) retraso mental leve o moderado; 2) anomalías esqueléticas; 3) talla baja; 4) alteraciones dermatoglíficas, y 5) facies característica, aunque se han descrito otras muchas anomalías presentes en un elevado porcentaje de pacientes con KS: paladar ojival/fisura palatina, cardiopatía congénita, alteraciones urogenitales, anomalías oftalmológicas. A pesar de su facies característica, con fisuras palpebrales largas, eversión del párpado inferior, cejas arqueadas y orejas grandes y despegadas, este síndrome no siempre es diagnosticado en nuestro medio. Presentamos 5 casos con rasgos característicos de KS que vinieron a nuestra consulta. Dos de ellos acudieron para consejo genético por presentar alteraciones oftalmológicas importantes (microftalmía, coloboma) acompañada de otras alteraciones. En uno de los casos se ha encontrado una duplicación parcial del cromosoma 9 pendiente de terminar de filiar. otros dos acudieron para descartar deleción del cromosoma 22 por presentar fisura palatina, uno para descartar síndrome de Prader-Willi por retraso mental, talla baja y obesidad. En estos casos, la presencia de ciertas anomalías importantes o de rasgos comunes a otros síndromes más conocidos y en los que la confirmación diagnóstica es posible, había enmascarado el diagnóstico de síndrome de Kabuki. La mayoría de los pacientes con KS tienen un cariotipo normal. Se han descrito, no obstante, varios casos con rasgos de KS asociados a diferentes anomalías cromosómicas sin puntos de rotura coincidentes y recientemente 6 casos con una duplicación que afectaba a la misma región (8p22-23). La existencia de un factor genético común en este grupo de pacientes abre la posibilidad de encontrar una etiología común del KS y la consiguiente creación de una prueba diagnóstica de confirmación, que facilitaría el diagnóstico.
Síndrome Hardikar: ¿una forma de presentación del síndrome de Kabuki? A propósito de un caso
I. Ejarque1, E. Piozzi2, R. Bono3 y F. Faravelli1
1Departamento Lígure de Genética Médica. Hospital Galliera. Laboratorio di Genetica Umana. Génova. 2Divisione Oculistica. Hospital Niguarda. Milán. 3Instituto Neurológico Besta. Milán. Italia.
Presentamos una paciente de 10 años con diagnóstico clínico de síndrome de Kabuki y con manifestaciones inusuales para este síndrome como hepatoblastoma y distrofia retiniana de tipo pigmentario. El síndrome de Hardikar, descrito en pocos pacientes, presenta características comunes al síndrome de Kabuki, y se caracteriza por estenosis/atresia congénita de las vías biliares intrahepáticas y distrofia retiniana. Discutimos si el síndrome de Hardikar pudiera representar un extremo del espectro clínico del síndrome de Kabuki a partir de la presentación del caso que presentó y que las complicaciones presentadas en nuestra paciente se deban encuadrar en este contexto.
Síndrome de Mehmo
A. González-Meneses López y G. Rodríguez Criado
Hospitales Universitarios Virgen Macarena y Virgen del Rocío. Sevilla. España.
Introducción: El síndrome de Mehmo (OMIM 300148) es un síndrome malformativo ligado al cromosoma X que se caracteriza por retraso mental, epilepsia, hipogonadismo, microcefalia y obesidad. Los pacientes descritos presentan un grado importante de retraso mental, con una baja esperanza de vida por o a causa de procesos respiratorios intercurrentes. El gen responsable se encuentra localizado entre Xp21-Xp22 aunque aún no ha podido ser aislado.
Caso clínico: Presentamos el caso de un niño nacido de un embarazo bien controlado, con parto a término por cesárea, y test de Apgar 8/10/10. Peso al nacer: 2.300 g; longitud, 46 cm, y perímetro craneal, 33 cm. No hay antecedentes familiares destacables. Al nacer se detectó hipotonía axial, hipogonadismo e hidrocefalia triventricular con microcefalia. A partir del quinto mes desarrolló obesidad e infecciones respiratorias frecuentes, con cierto grado de espasticidad. Se le puso una derivación ventriculoperitoneal por la hidrocefalia. Con un año y medio de edad desarrolló crisis convulsivas con hipsarritmia en el electroencefalograma (EEG). Actualmente tiene 2 años de edad, con un importante retraso que empeoró desde la aparición de las crisis convulsivas. A la exploración destaca microcefalia con talla baja (< P3) pero con obesidad (peso en P75), telecanto, importante hipotonía axial y leve espasticidad de los miembros. Las hendiduras palpebrales se encontraban hacia arriba, con carrillos llenos y orejas con lóbulos de la oreja carnosos y algo plegados, con las comisuras de la boca hacia abajo. Discreto edema de manos y pies, estos últimos en equino. Los genitales son hipoplásicos con testes en el canal inguinal e hipospadias balánica. Pruebas complementarias: hemograma y bioquímica básica, pruebas de función hepática, hormonas tiroideas y de crecimiento, normales. Acidosis respiratoria que coincidía con los problemas respiratorios, sin acidosis metabólica; creatinfosfocinasa, colesterol y triglicéridos normales; lactato deshidrogenasa, levemente elevada. Cariotipo y estudio de Prader-Willi, normales. EEG: hipsarritmia. RM craneal: atrofia corticosubcortical con dilatación ventricular.
Conclusiones: Les comunicamos un nuevo caso de síndrome de Mehmo. El diagnóstico diferencial debe realizarse fundamentalmente con otros procesos que combinan microcefalia, obesidad, hipogenitalismo, convulsiones y retraso mental, fundamentalmente el síndrome de Borjeson-Forssman-Lehman, también ligado al cromosoma X.
Variante de síndrome de Axenfeld-Rieger. Caso clínico
V.M. Martínez González1, I. Lorda-Sánchez1, P. Ruiz Barnes2, M.J. Trujillo Tiebas1, M. García Hoyos1, R. Riveiro Álvarez1, C. Ramos1 y C. Ayuso1
1Servicio de Genética. 2Servicio de Neurología. Fundación Jiménez Díaz. Madrid. España.
Introducción: El síndrome de Axenfeld-Rieger (RIEG) agrupa un amplio espectro fenotípico en el que se incluyen anomalías oculares (anomalía de Axenfeld-Rieger [ARA]), acompañadas de signos sistémicos (síndrome de Rieger). La ARA se caracteriza por embriotoxón posterior, iridogoniodisgenesis, corectopia, etc., y conduce a glaucoma en el 50 % de los casos. Los signos extraoculares del RIEG son dientes pequeños o ausentes, piel periumbilical redundante, pérdida auditiva sensorial, defectos cardíacos y dismorfismo craneofacial leve. El patrón hereditario es autosómico dominante con penetrancia incompleta y expresividad variable para los signos oculares y extraoculares. Para el RIEG se han encontrado dos genes relacionados con el desarrollo ocular que codifican para factores de transcripción, PITX2 (pituitary homeo box 2) y FOXC1 (forkhead box transcription factor C1), además de al menos dos loci genéticos, 13q14 (RIEG2) y 16q24, cuyos genes aún no se han caracterizado. Mutaciones en el PITX2 (4q25) causan el RIEG tipo 1 (RIEG1) con una fuerte relación genotipo-fenotipo, a diferencia del gen FOXC1 (6p25), con el que no se puede predecir la intensidad y compromiso fenotípico por el tipo de alteración molecular.
Caso clínico: Mujer de 22 años que al nacimiento presentó pie equinovaro derecho, facies dismórfica e hipertelorismo; a los 4 años se diagnostica estrabismo, glaucoma y osteocondritis femoral bilateral con aplanamiento de epífisis, así como hidrocefalia tratada a los 5 años de edad. Es remitida a genética con un informe oftalmológico de ARA con epicanto, corectopia, embriotoxón anterior y sinequias iridianas anteriores periféricas; al examen se aprecia leve retraso psicomotor, obesidad generalizada y una posible sordera leve. El padre muestra algunos rasgos dismórficos faciales (oculares) similares, sin ninguna otra alteración ni retraso psicomotor.
Discusión y conclusiones: DeHauwere et al (1973) y Chitty et al (1991) describen 2 casos familiares de anomalía de Axenfeld-Rieger con manifestaciones extraoculares como: hipertelorismo, hipoplasia mediofacial, sordera sensorial leve, hidrocefalia, retraso psicomotor y epífisis femorales aplanadas. Los autores proponen que estas características conforman un síndrome separado. El caso presentado concuerda con lo aportado por estos autores, siendo así el tercer caso descrito de esta entidad en la que sus límites sindrómicos con el RIEG, así como su etiopatogenia molecular, están aún por definir.
Deleción terminal 18q: una causa de desmielinización
I. Arroyo Carrera1, A. López Lafuente1, P. Rodríguez Santano1, M.J. García García1, V. Carretero Díaz1 y L. Rodríguez Martínez2
1Servicio de Pediatría. Hospital San Pedro de Alcántara. Servicio Extremeño de Salud. Cáceres. 2ECEMC, Centro de Investigación sobre Anomalías Congénitas (CIAC). Instituto de Salud Carlos III. Madrid. España.
Introducción: La deleción terminal 18q es un síndrome de macrodeleción terminal con retraso mental y malformaciones que tiene su base en la haploinsuficiencia de múltiples genes, entre los cuales suele encontrarse el gen para la proteína básica de la mielina (PBM) con locus en 18q23. El fenotipo presenta una gran variabilidad y se caracteriza por retraso mental y de crecimiento, hipoacusia, anomalías faciales, genitales y de los pies. Esta variabilidad podría estar relacionada con la existencia de otros factores (genes moduladores a distancia, ambientales) además de la monosomía de los genes implicados en la deleción.
Presentamos 2 casos clínicos con deleción terminal 18q con diferente expresividad clínica, uno de ellos muy intenso, y el otro muy poco expresivo facialmente cuyo diagnóstico fue un hallazgo casual en el cariotipo. Ambos casos, con deleción del locus para la PBM, presentan en la RM cerebral signos de desmielinización.
Casos clínicos:Caso 1. Gemelo dicoriónico producto de fertilización in vitro, a término, sin antecedentes de interés, cogemelo sano. Gran expresividad clínica al nacimiento con genitales ambiguos (pene pequeño, 18 mm; hipospadias penoescrotal, bolsa escrotal derecha hipoplásica con menor rugosidad cutánea sin teste palpable), frente amplia con fontanela anterior grande, hipoplasia mediofacial, boca en carpa, filtro plano, orejas de implantación baja y uñas de los pies hipoplásicas. Evolutivamente presentaba retraso mental grave y de crecimiento con sordera neurosensorial más alteración conductual. Caso 2. Niña de 10 años remitida para valoración por retraso psicomotor con sordera mixta detectada a los 4 años de edad. Se había realizado cirugía para ampliación de CAES, llevaba prótesis auditivas. Talla límite (P3-10). Fenotipo facial no característico con hipertelorismo. Dentro del estudio diagnóstico que incluyó cariotipo y resonancia cerebral se encontró la anomalía cromosómica.
Cariotipos de alta resolución (550-850 bandas): primer caso, 46,XY, del(18)(q23-qter). ish del(18) (q23)(D18S1390-). Segundo caso 46,XX, del(18)(q22.2-qter). ish (tel 18q-). Cariotipos de los padres de ambos casos normales.
En las resonancias cerebrales de los 2 pacientes se observan signos de desmielinización.
Comentario: La diferente expresión clínica que presentan nuestros 2 pacientes, más intensa en el que presenta la deleción más terminal, concuerda con los datos de la literatura médica donde no se puede establecer claramente una correlación fenotipo/genotipo, pero que sí parece identificar la región crítica del síndrome en la banda terminal 18q23. El estudio mediante FISH con sonda para la región telomérica del brazo largo del cromosoma 18 permite confirmar la deleción terminal, no intersticial, de nuestros casos con pérdida del locus para la PBM. Se ha demostrado en series de pacientes que todos los que tienen delecionada una copia del locus para la PBM presentan alteraciones de la sustancia blanca en la RM, siendo normal en aquellos con deleciones intersticiales que conservan el locus, pudiendo interpretarse esta alteración como secundaria a la haploinsuficiencia de la PBM.
Conclusión: Debe incluirse a la deleción terminal 18q entre las causas de desmielinización y ser tenida en consideración en el diagnóstico diferencial de estos pacientes, sobre todo para buscar posibles manifestaciones clínicas del cuadro en los pacientes con menos expresión clínica y de más difícil diagnóstico.
Displasia oculodentodigital: expansión del fenotipo
B. Gener1, A. Ortiz2, M. Lizarraga2, R. Martínez2 y E.W. Jabs3
1Unitat de Genètica, Universitat Pompeu Fabra. 2Hospital de Cruces. Baracaldo. Vizcaya. España. 3Institute of Genetic Medicine. The Johns Hopkins Hospital. Baltimore. Estados Unidos.
La displasia oculodentodigital (ODDD; OMIM 1642001) es una entidad que afecta al desarrollo de la cara, los ojos, las extremidades y la dentición. Se hereda de forma autosómica dominante, tiene una alta penetrancia y gran variabilidad en la expresión. Se presenta el caso de un padre y su hijo con unos rasgos faciales característicos consistentes en una nariz fina con hipoplasia de las alas nasales, hendiduras palpebrales pequeñas y anomalías de la dentición asociadas a sindactilia completa del cuarto y quinto dedo en ambas manos con agenesia de la segunda falange del quinto dedo bilateral y presencia de hipospadias glandular. El niño presenta un discreto retraso psicomotor y en el padre la presencia de vejiga neurógena se explica por la posible aparición de este trastorno en la segunda década de la vida. Sabemos que otros miembros de la familia paterna presentan rasgos faciales o anomalías en las manos de forma aislada, aunque no han podido ser valorados por residir en otro país. Recientemente se han descrito, en 17 familias diagnosticadas de ODDD, la presencia de mutaciones, predominantemente de cambio aminoacídico en el gen GJA1 localizado en 6q22-q24 que codifica para la conexina 43. La presencia de mutación en este gen también se ha podido comprobar en nuestros pacientes. Si bien otras anomalías se han asociado ocasionalmente a ODDD, la asociación con hipospadias no ha sido previamente comunicada. Es de esperar que próximos estudios del gen responsable de la ODDD permitan dilucidar su potencial intervención en el desarrollo del sistema genitourinario.
Variabilidad fenotípica en una familia de tres generaciones con síndrome craneofacial sordera mano
M.D. Saavedra-Ontiveros, P. Blanco y M. Orera Clemente
Unidad de Genética. Hospital General Universitario Gregorio Marañón. Madrid. España.
Introducción: El síndrome craneofacial-sordera-mano (CDHS, OMIM 122880) comparte características con el síndrome de Waardenburg I (WSIII) como el telecanto, la hipoplasia de alas nasales y la hipoacusia. Al igual que WSIII presenta alteraciones de las extremidades caracterizadas por desviación cubital de ambas manos. Se hereda en forma autosómica dominante y se ha descrito un grupo familiar con una mutación sin sentido en el gen PAX3.
Caso clínico: Varón de 6 años hijo único de padres no consanguíneos con facies triangular, telecanto, hipoplasia de alas nasales, labio superior fino, pabellones auriculares pequeños, de implantación baja y ligeramente displásicos, que presenta marcada desviación cubital de ambas manos, sindactilia cutánea incompleta y camptodactilia del dedo medio. En la audiometría se hizo patente una hipoacusia moderada. Su padre presenta hipoplasia de alas nasales y pabellones auriculares pequeños y su abuela paterna tiene un mechón de cabello blanco desde la infancia.
Discusión: Las características del paciente sugieren la presencia de un síndrome craneofacial sordera-mano, mientras que el padre y la abuela podrían representar variantes fenotípicas leves del WSII. A pesar de que no se ha realizado todavía el estudio molecular de la familia, es de suponer que los miembros afectados tengan, todos, la misma mutación. En este caso las diferencias fenotípicas observadas serían debidas a diferentes complementos genéticos modificadores de la penetrancia. Todo ello nos hace postular que el síndrome craneofacial-sordera-mano representa una variante del espectro hipopigmentación-sordera que constituyen las distintos tipos del síndrome de Waardenburg.
Síndrome de Van der Woude. A propósito de 4 casos dentro de una misma familia
J. Guerrero-Fernández, M.T. García-Ascaso, D. Plaza López de Sabanda, M.A. Molina Rodríguez, A. Alcalde de Alvaré y R. Gracia Bouthelier
Servicio de Endocrinología Pediátrica. Hospital infantil La Paz. Madrid. España.
El síndrome de Van der Woude o de las fositas labiales representa una entidad hereditaria rara aunque de muy fácil diagnóstico y cuyo conocimiento sería preciso para un adecuado consejo genético. Presentamos el caso de un niño de 2 años de edad que nos remitieron para valoración de criptorquidia. En la exploración clínica descubrimos dos pequeñas eminencias coronadas, cada una, por una leve depresión y localizadas de forma simétrica y centrada en labios inferiores. En los antecedentes personales destacaba haber sido intervenido de fisura palatina; con respecto a los familiares, la hermana presentaba las mismas lesiones (fositas labiales con fisura palatina intervenida), así como el padre y el abuelo paterno (exclusivamente fositas labiales). El interés del caso radica en la importancia que hubiese tenido el consejo genético por cuanto se trata de un proceso heredable de forma autosómica dominante, de elevada penetrancia y expresividad variable. Relacionado con esto último, las malformaciones posibles dentro de una misma familia incluyen, fundamentalmente, la existencia de fositas labiales, fisura labiopalatina e hipodontia. Las mutaciones encontradas hasta la fecha se localizan a nivel de 1q32-41 (forma clásica), 1p34 (tipo 2) y 17p11.2-11.2 (forma modificada).
Síndrome de Floating-Harbor
M.A. Molina Rodríguez, A. Alcalde de Alvaré, J. Guerrero-Fernández y R. Gracia Bouthelier
Servicio de Endocrinología Pediátrica. Hospital infantil La Paz. Madrid. España.
Presentamos 2 niños varones de 4 y 16 años no relacionados entre si con las típicas características del síndrome de Floating-Habor: rasgos craneofaciales (cara triangular, nariz bulbosa con puente nasal alto, orificios nasales grandes, ojos grandes pero hundidos, boca ancha con labios finos y orejas retrovertidas); talla baja proporcionada (4 DE) con edad ósea retrasada, clinodactilia del quinto dedo de la mano y cuello corto; retraso en el área del lenguaje expresivo. Ambos pacientes fueron de bajo peso y talla al nacer con disminución progresiva de la velocidad de crecimiento. Las tallas parentales son normales en un caso pero en otro es de 3 DE. Se han descartado enfermedad celíaca y endocrina, siendo el cariotipo 46XY masculino normal. En un paciente se ha iniciado tratamiento con hormona de crecimiento (GH biosintética). Se han descrito unos 24 casos de síndrome de Floating-Harbor. La etiología es desconocida. Se trata de casos esporádicos dentro de familias normales. En 3 casos se sugieren herencia autosómica dominante o variante familiar, basado en rasgos peculiares maternos.
Síndrome de Costello. Un síndrome de pronóstico muy difícil en la lactancia
L. Toledo Bravo de Laguna1, M. Martí Herrero1, J.C. Cabrera López1, C. Trujillo Cabrera1 y M. Del Campo Casanelles2
1Unidad de Neurología Infantil. Hospital Materno-Infantil. Las Palmas. 2Unidad de Genética. Departamento de Ciencias Experimentales y de la Salud. Universidad Pompeu Fabra. Barcelona. España.
Se presenta el caso de un niño de 5 meses diagnosticado intraútero de linfangioma cervical. Tuvo un parto prematuro con peso elevado para su edad gestacional. Presentaba un fenotipo especial asociado a una estenosis valvular pulmonar grave, ectasia pielocalicial bilateral y problemas importantes para la alimentación y ganancia de peso. Tras establecer el diagnóstico clínico de síndrome de Costello, reevaluamos el caso de un segundo lactante, inicialmente diagnosticado de síndrome progeriforme, que presentaba unas características clínicas muy similares y que falleció por su cardiopatía.Costello describió en 1970 los casos de 2 pacientes con elevado peso al nacer, dificultades para la alimentación en el período neonatal, retraso mental y un fenotipo físico muy característico; posteriormente se aceptó que el anteriormente descrito síndrome facio-cutáneo-esquelético era la misma entidad. La herencia es autosómica dominante y la mayoría de los casos son de novo. Aunque no se conoce el defecto genético subyacente, se ha observado una alteración en la síntesis de las fibras de elastina por defecto de la proteína BDP (elastin-binding protein). El diagnóstico clínico se establece mediante la identificación de los siguientes hallazgos: elevado peso al nacer, dificultades para la alimentación a veces muy graves, retraso mental, macrocefalia, pelo rizado, cara tosca con frente amplia, con hipertricosis, hendiduras palpebrales descendentes, orejas grandes, rotadas, con lóbulos grandes y de implantación baja, labios gruesos, nariz bulbosa de base ancha con narinas antevertidas, hiperlaxitud articular, sobre todo en los dedos, falanges distales anchas y posición anómala de los pies (talo vertical). La piel es laxa y redundante, sobre todo en el cuello y dorso de manos y pies. Los surcos palmares y plantares son muy profundos. Suelen aparecer hiperqueratosis y papilomas en nariz o en otras localizaciones. Además de anomalías cardíacas congénitas y del SNC, es importante reconocer que puede existir un riesgo incrementado de tumores malignos.
Detección de anomalías cromosómicas subteloméricas en pacientes con retraso mental de etiología no determinada en el sureste de Escocia
S. García-Minaur, P. Taladianou, E. Maher, W. Teik Keng, W.K. Lam, E.M. Porteous y D.R. FitzPatrick
South East of Scotland Genetics Service. Western General Hospital. Edimburgo. Reino Unido.
Se presentan los resultados del programa de detección de anomalías cromosómicas subteloméricas en el sureste de Escocia. Los criterios de inclusión se establecieron de antemano y comprenden la presencia de antecedentes familiares, rasgos clínicos específicos y determinadas investigaciones previas. Todos los pacientes fueron evaluados personalmente por un genetista clínico. Durante un período de 17 meses (de enero de 2002 a mayo de 2003) se estudiaron 177 pacientes con retraso mental de etiología no determinada. En 6 casos se detectó una deleción visible microscópicamente, previo al empleo de técnicas moleculares para estudio de anomalías subteloméricas. En otros 6 casos se conocía con antelación la presencia de una anomalía cromosómica, pero se consideró que esta no justificaba el fenotipo (3 translocaciones aparentemente equilibradas, 2 aneuploidías de cromosomas sexuales y una inversión paracéntrica). El programa de detección encontró anomalías subteloméricas en 4 casos (una deleción y 3 translocaciones no equilibradas). En uno de ellos, un progenitor resultó ser portador de una translocación equilibrada críptica. En los 3 casos restantes la anomalía era de novo. La frecuencia total de anomalías cromosómicas en la población en estudio fue de 5,6 %. La frecuencia de anomalías subteloméricas fue 2,3 %, inferior a la comunicada en otras series. Es posible que la alta calidad de los estudios citogenéticos y la selección de pacientes expliquen esta aparente baja tasa de detección.
Importancia del estudio citogenético de alta resolución para el diagnóstico de anomalías cromosómicas no balanceadas. A propósito de 50 casos
E. Galán Gómez1,2, J.M.ª Carbonell Pérez2, J. Sáenz Hurtado2, J.E. Arroyo Moñino1, J. Ramón Sicilia2, M.ªC. Ledesma Alcázar2, N. Amaya Corchuelo2, R. Gallardo Gutiérrez 2, E. Julián Polo2, F.J. Lara Laranjeira2, G. Rodríguez Criado3, F.J. Ramos Fuentes4, I. Bueno Martínez4 y J.J. Cardesa García1
1Departamento de Pediatría. Hospital Materno-Infantil. Facultad de Medicina. UEX. Badajoz. 2Unidad de Genética. Centro Extremeño de Desarrollo Infantil. Hospital Perpetuo Socorro. Badajoz. 3Unidad de Dismorfología. Hospital Virgen del Rocío. Sevilla. 4Facultad de Medicina. Universidad de Zaragoza. España.
Hemos revisado los estudios cromosómicos de alta resolución que realizamos en nuestro laboratorio de citogenética durante el período de 1/1/2000 a 31/12/2003. Se han realizado 1.424 cariotipos de alta resolución en sangre periférica. De ellos, 74 (5,2 %) presentaban una alteración cromosómica estructural, 50 casos eran anomalías cromosómicas desequilibradas y 24 presentaban anomalías cromosómicas balanceadas. Los casos que incluimos son aquellos que fueron diagnosticados al microscopio por alta resolución. Excluimos aquellos pacientes que tenían algún síndrome de microdeleción, cuyo estudio de alta resolución fue normal, y posteriormente fueron diagnosticados por técnicas de citogenética molecular. La técnica utilizada fue la técnica de bandas GTG de alta resolución de bloqueo de timidina y posterior liberación con con 2-desoxicitidina modificada de Wheater y Roberts (1987). El nivel de bandas utilizado osciló entre 550 y 850 bandas.
De los 50 casos patológicos con anomalías cromosómicas no balanceadas, 24 casos tenían realizado uno o más cariotipos previos que fueron normales; 10 casos tenían un cariotipo previo cuyo estudio había sido incompleto; 2 casos tenían cariotipos previos patológicos que eran erróneos y 14 casos no tenían realizado un cariotipo previo. Las anomalías encontradas fueron 25 microdeleciones, 14 duplicaciones, 3 duplicaciones más deleciones, 4 marcadores, 2 isocromosomas y 2 anillos. De los pacientes patológicos que tenían cariotipos previos de resolución estándar normales, 16 correspondían a deleciones, 7 a duplicaciones y uno presentaba una duplicación y una deleción. Las microdeleciones afectaron con más frecuencia a los cromosomas 2q (4 casos), 4p (4 casos), 17p (3 casos) y 18p (3 casos). Las duplicaciones afectaron con mayor frecuencia a 2q (2 casos), 8p (2 casos) y 9p (2 casos).
Todos los casos fueron valorados por un genetista clínico. El estudio cromosómico de alta resolución fue indicado en 39 casos por el genetista clínico, en 4 casos por el neurólogo infantil, en 4 casos por el pediatra y en 3 casos por otros médicos. En 44 pacientes, la indicación del estudio fueron rasgos dismórficos y/o retraso mental. En 7 casos el resultado citogenético fue concordante con la sospecha clínica.
Con este estudio se desea señalar la importancia de la realización del estudio cromosómico de alta resolución en pacientes con rasgos dismórficos y/o defectos congénitos y/o retraso mental, aun en pacientes que tuvieran realizado uno o más cariotipos de resolución estándar previos con resultado normal.
Las deleciones menores (1,5-2 Mb) son más frecuentes en los casos familiares que en los casos aislados de síndrome de microdeleción 22q11.2
L. Fernández1, P. Lapunzina1, I. López Pajares1, G. Rodríguez Criado3, L. García Guereta2 y A. Delicado1
Hospital Universitario La Paz, 1Servicio de Genética Médica. 2Servicio de Cardiología Infantil. 3Servicio de Dismorfología. Hospital Virgen del Rocío. Sevilla. España.
Introducción: El síndrome de microdeleción 22q11.2 tiene una frecuencia en la población general de 1 cada 4.000-6.000 nacidos vivos. Se ha observado una deleción común mayor (3 Mb) en el 90 % de los casos, una deleción común menor (1,5-2 Mb) en el 8 %, y deleciones atípicas en el 2 %.
Material y métodos: Se han realizado por hibridación in situ fluorescente (FISH) análisis de la región 22q11.2 aplicando las sondas locus específicas D22S75 y TUPLE1. Los estudios moleculares se realizaron mediante la amplificación por PCR de 12 a 16 marcadores polimórficos de la región 22q11.2. De los 59 pacientes con deleción 22q11.2 diagnosticados por FISH, 6 (10 %) resultaron ser casos familiares. De ellos, cinco se han estudiado molecularmente junto con sus familiares (total 19 individuos).
Resultados: Los resultados citogenéticos y moleculares confirman en todos los casos una herencia materna de la deleción. En una familia con tres generaciones de afectados, la deleción fue transmitida por el abuelo materno del propósito. Por genotipificación de microsatélites se identificó una familia con deleción común mayor (3 Mb) y 3 familias con deleción común menor (1,5-2 Mb). En otra familia, la ausencia de datos parentales no permitió definir el tamaño de la deleción.
Conclusiones: Los resultados sugieren un predominio de la deleción común menor en 3/4 de los casos familiares (75 % frente a 8 % de la población general delecionada). Esto apoyaría la hipótesis de una mayor tolerancia de las deleciones más pequeñas en los casos heredados. La presencia de una deleción común pequeña (1,5-2 Mb) o atípica obligaría a descartar segregación familiar del síndrome.
Síndrome de Beckwith-Wiedemann con duplicación 11p15.5 de origen paterno asociada a síndrome de Klinefelter e inversión pericéntrica del cromosoma Yde novo
M. Palomares1, A. Delicado1, P. Lapunzina1, M.A. Molina2, E. Galán Gómez3 e I. López Pajares1
1Servicio de Genética Médica. Hospital Universitario La Paz. Madrid. 2Servicio de Endocrinología Infantil. Hospital Universitario La Paz. Madrid. 3Unidad de Genética. Hospital Materno-Infantil. Badajoz. España.
Caso clínico: Primer hijo de padres sanos y no consanguíneos. Durante el embarazo se observó polihidramnios por cuyo motivo se realizó estudio citogenético prenatal detectándose un cariotipo fetal: 47,XX,inv(Y)(p11.2 q11.23) de novo. Parto a las 31 semanas con peso al nacimiento de 2.050 g (> percentil 97), talla 42 cm (P50-75) y perímetro craneal de 29 cm (P50). En la exploración se observó dismorfia facial con macroglosia, orejas de implantación bajas, hipertelorismo, epicanto e hipoplasia facial media. Además, el niño presentaba una criptorquidia bilateral asociada hipospadias, hernia umbilical y ano desplazado anteriormente. La exploración neurológica mostraba una hipotonía generalizada. La TC reveló una hemorragia intraventricular y disgenesia del cuerpo calloso. Ecografía renal: ureterohidronefrosis izquierda. Evolución: a los 15 meses comienza con convulsiones asociadas a alteraciones en el electroencefalograma (EEG). A 21/2 años de edad es intervenido de criptorquidia, hernia umbilical y malformación renal. A los 3 años fue reevaluado por presentar retraso mental y dismorfia facial, y se estableció el diagnóstico clínico de probable síndrome de Beckwith-Wiedemann (SBW). Estudios citogenéticos: el cariotipo del paciente se repitió a la edad de 4 años. Se realizaron técnicas de hibridación in situ fluorescente (FISH) utilizando la sonda para región subtelomérica de brazos cortos del cromosoma 11 (D11S2071, Vysis). Se analizaron 20 metafases y se observaron tres señales en todas las células examinadas. Dos señales fueron localizadas en brazos cortos de ambos cromosomas 11 y una tercera señal adicional estaba situada en la porción distal de los brazos largos de un cromosoma 18. El estudio citogenético de los padres mostró que el padre del paciente era portador de una translocación críptica balanceada entre los brazos cortos de un cromosoma 11 y los brazos largos de un cromosoma 18. Su cariotipo era: 46,XY, t(11;18)(p15.5;q23). ish t(11;18) (p15.5; q23)(D11S2071-;D11S2071 +). El cariotipo materno fue normal. Así pues, el niño había heredado el cromosoma derivado 18 paterno, presentando una duplicación de la región 11p15.5 → pter asociada a una deficiencia de la región 18q23 → qter, siendo su cariotipo 47,XX, inv(Y)(p11.2q11.2), der(18)t(11;18)(p15.5;q23) pat.ish der (18) t(11;18)(p15.5;q23) (D11S2071+). Estudios moleculares: se realizaron análisis de marcadores polimórficos de la región 11p15.5, del cromosoma X y del cromosoma Y. En la región 11p15 uno de los microsatélites (D11S4088) mostró un patrón de 3 alelos señalando su triplicación pero no siendo informativo para identificar el origen parental del material 11p extra. Dos de los marcadores del cromosoma X (DX8091, DXS1214) resultaron informativos revelando que el cromosoma X extra era de origen paterno. Finalmente, el análisis de los marcadores del cromosoma Y no mostró deleción en el cromosoma Y reestructurado. Conclusiones: sólo un 2-3 % de los pacientes con SBW presentan anomalías cromosómicas en la región 11p15.5. Los hallazgos clínicos en pacientes con el SBW debido a trisomías 11p15.5 ponen de manifiesto una mayor incidencia de retraso mental respecto a los pacientes con cromosomas normales. Por ello, creemos necesario realizar estudio citogenético molecular (FISH) en todos los pacientes con SBW, dado que el estudio citogenético convencional puede no ser efectivo en la detección de pequeñas reestructuraciones en desequilibrio. Esto permitirá identificar familias con reestructuraciones en balance con gran riesgo de tener hijos afectados en posteriores embarazos.
Caracterización molecular de un cromosoma marcador implicando 6cen6p11.2 en un niño con microcefalia y craneosinostosis
O. Villa1, M. Del Campo2, M. Salido1, L. Astier1, J. del Valle2, L. Pérez-Jurado2 y F. Solé1
1Laboratorio de Citogenética y Biología Molecular. Servicio de Patología. Hospital del Mar. Barcelona. 2Unidad de Genética. Universitat Pompeu Fabra. Barcelona. España.
Introducción: La trisomía parcial de la región 6p es muy poco frecuente. Se han descrito unos 40 casos que afectan a la región más terminal de este cromosoma. Presentamos un caso con trisomía parcial de una región más centromérica (6p11.2 → 6cen) en forma de cromosoma adicional.
Caso clínico: Recién nacido varón hijo de madre de 36 años sin antecedentes familiares de interés, embarazo de curso normal y parto eutócico con peso y talla al nacer en el P50. Al nacer se observa perímetro craneal 32,5 cm en P10 y con braquicefalia, plagiocefalia occipital asimétrica, fontanela mayor no palpable y suturas con signos de fusión prematura. La presencia de craneosinostosis incipiente global se confirmó en la radiografía de cráneo. Presentaba asimismo una mínima hipoplasia ungueal. El desarrollo psicomotor del paciente ha sido normal hasta los 10 meses de vida. Estudio citogenético: el estudio citogenético convencional a partir de sangre periférica reveló la presencia de un cromosoma marcador de pequeño tamaño, con fórmula cromosómica inicial de 47,XY, + mar [13]/46, XY [7]. Los cariotipos de los padres eran normales. Para caracterizar la procedencia del fragmento se aplicó primero la técnica de SKY-FISH, que lo identificó como material procedente del cromosoma 6. Este resultado se comprobó mediante pintado cromosómico específico para este cromosoma. Para poder precisar la región afectada se realizó la técnica de hibridación genómica comparada (CGH), que reveló ganancia desde la región centromérica hasta 6p11.2, quedando la fórmula cromosómica del siguiente modo: 47,XY, + mar, ish mar CEP6+, WCP6+. Se realizó un estudio molecular con tres marcadores microsatélites, localizados en 6q15, 6p11.2 y en 6p21.2. La presencia de tres alelos en el locus 6p11.2 confirmó la existencia de una duplicación de origen materno. Se está llevando a cabo el mapeo de la región implicada mediante hibridación in situ fluorescente (FISH) con BAC.
Discusión: Este caso ilustra la necesidad de solicitar un cariotipo en presencia de una microcefalia aislada con craneosinostosis. En ausencia de posibles factores extrínsecos con efecto deformativo, la craneosinostosis puede ser secundaria a una microcefalia primaria o bien estar causada por una anomalía ósea primaria. No existe ningún gen causante de craneosinostosis identificado hasta el momento en la región duplicada.
Agradecimientos: Red de Centros de Genética (G03/07); Red de Centros de Cáncer (C03/10); Vicens Catalá (Prenatal Genetics).
Implicaciones clínicas y caracterización de un cromosoma marcador detectado prenatalmente
J.M. Belloso1, M. Roselló2, E. Gabau3, N. Baena1, F. Mellado3, M.R. Caballín4 y M. Guitart1
Unidad de Diagnóstico Prenatal. Servicios de 1Laboratorio, UDIAT-Centro Diagnóstico, 2Pediatría y 3Servicio de Obstetricia y Ginecología. Corporación Sanitaria Parc Taulí. 4Unidad de Antropología. Facultad de Ciencias de la UAB. Sabadell. Barcelona. España.
Los cromosomas extras se presentan con una frecuencia de 0,4-1,5 en la amniocentesis y crean gran dificultad en el consejo genético, dado el variado abanico de posibilidades fenotípicas que puede presentar el feto. Las técnicas citogenéticas convencionales, en general, no permiten conocer el origen de estos marcadores, por lo que es necesario recurrir a otras técnicas de diagnóstico genético como hibridación in situ fluorescente (FISH) e hibridación genómica comparada (HGC). Se presenta un caso de un niño nacido de una segunda gestación de padres no consanguíneos y sin antecedentes de interés. Al detectarse en el primer trimestre de embarazo translucencia nucal, se realizó una amniocentesis precoz y se observó en el cariotipo fetal un cromosoma marcador extra en el 80 % de las metafases cuya presentación fue de novo. Las bandas C mostraron una región heterocromática correspondiente al centrómero, y una región eucromática, y las bandas Q no mostraron satélites. Al aplicar las sondas centroméricas de los cromosomas 3, 13, 14, 22, 18, X e Y y las sondas específicas de locus de 15q12 y 22q11.2 no se pudo caracterizar el cromosoma marcador. Los padres decidieron seguir el embarazo al no presentar malformaciones asociadas. El embarazo llegó a término y en el recién nacido se observó una leve dismorfia facial, piel sobrante en la nuca y orejas grandes. En la última exploración física del niño, con una edad de 3 años y 6 meses, destaca: unas hendiduras palpebrales antimongoloides, plenitud periorbitaria, orejas grandes y displásicas, cuello corto, piel sobrante en nuca, implantación baja de cabello tanto anterior como posterior y tronco alargado. Desde el punto de vista neurológico presenta un trastorno específico del lenguaje, sobre todo en el aspecto fonológico, una conducta hiperactiva y trastorno del sueño. Tras la puesta a punto de la técnica de HGC en nuestro laboratorio, se aplicó a una muestra de sangre periférica del paciente observándose una ganancia de 8p10-p12. Este resultado se confirmó mediante pintado cromosómico del cromosoma 8 y sonda centromérica del mismo. La técnica de HGC constituye una buena alternativa para conocer el origen de los cromosomas marcadores, no caracterizados con los métodos convencionales. Si bien nuestro paciente presenta algunas características físicas externas compatibles con la trisomía 8p, debido al pequeño tamaño del cromosoma extra, es difícil establecer una estrecha correlación genotipo-fenotipo.
Presentación de 2 casos con una deleción 14q proximal
F. López-Grondona1, E. Mansilla1, L. Rodríguez1 y M.L. Martínez-Frías1,2
1CIAC. Instituto de Salud Carlos III. Ministerio de Sanidad y Consumo. 2Departamento Farmacología. Facultad de Medicina. Universidad Complutense. Madrid. España.
Introducción: Con el aumento de la resolución en los cariotipos, a niveles que superen las 550 bandas, se han logrado visualizar alteraciones cromosómicas difíciles de detectar con menos resolución. Presentamos 2 casos que tienen una deleción observable sólo con cariotipo de alta resolución.
Casos clínicos:Caso 1. Lactante varón de 7 meses de edad, cuyos padres consultan a nuestro centro para asesoramiento genético. Antecedentes: segundo hijo de padres no consanguíneos, madre sana de 33 años y padre de 34 con antecedentes de epilepsia secundaria a traumatismo, con crisis desde los 9 meses hasta los 7 años, tratamiento antiepiléptico hasta los 18 años, y secuela de hemiparesia derecha. La hija anterior tiene 3 años y es sana. La gestación fue controlada, normal, sin patología materna y sin exposición a teratógenos. El parto: por cesárea a las 38 semanas, debido a una cesárea en el embarazo anterior. Peso al nacimiento en el P75-90, talla en el P75-90 y perímetro cefálico en el P50-75. Test de Apgar 9-10. Al sexto mes de vida se observa microcefalia y retraso en adquisiciones motoras, episodios sugerentes de espasmos infantiles poco definidos, con talla y peso conservados. Los estudios complementarios mostraron RM cerebral con agenesia parcial de cuerpo calloso, electroencefalograma (EEG) (2), normales, y cariotipo 46,XY, normal. En el examen físico a los 7 meses presentaba peso en el P75-90, talla en P50, y perímetro craneal (PC) por debajo del P3. Cráneo con aplanamiento occipital y leve trigonocefalia. Clinodactilia del quinto dedo de ambas manos, tono aumentado e irritabilidad fácil, sostén cefálico aún no consolidado, no fija la mirada ni sigue objetos. Comienza un programa de estimulación Vojta. A los 7 meses y 2 semanas tuvo que ser ingresado por presentar un episodio paroxístico afebril, con estudio metabólico, analítica y EEG normales. Cariotipo de alta resolución 46,XY, del(14)(q12;q2.11) de novo. Caso 2. Niña de 2 años y 2 meses que acudió a nuestro centro con la hipótesis diagnóstica de síndrome de Aicardi. Antecedentes: primera hija de madre de 33 años, operada a los 30 años de bocio nodular y en tratamiento desde entonces con Levothroid, eutiroidea y controlada durante la gestación. Padre 33 años sano; no hay consanguinidad. Embarazo controlado, sin patologías. Parto espontáneo a las 38 semanas. Peso de nacimiento en el P50, talla al nacimiento en el P25 y perímetro cefálico en el P25. Presentaba clinodactilia del cuarto dedo del pie derecho. Manifiesta dificultad respiratoria nasal, con cianosis y espiración ruidosa, por lo que permanece 5 días en cámara hood. Fue dada de alta con 11 días y diagnóstico de insuficiencia respiratoria neonatal autolimitada. Evoluciona con pobre ganancia ponderal, e ingresa para estudio a las 10 semanas de vida con un peso por debajo del P3, talla en el P50, y perímetro cefálico menor del P3. Evoluciona con microcefalia, dificultad alimentaria con medro pobre, estreñimiento, hipotonía y retraso psicomotor. Se realiza TC que muestra agenesia de cuerpo calloso e imágenes de aumento del espacio subaracnoideo frontal bilateral. Cariotipo convencional 46,XX normal. Realiza tratamiento de rehabilitación sin que la madre note progresos significativos. Por un fondo de ojo que sugiere coriorretinitis se plantea el síndrome de
Aicardi, diagnóstico que se descarta posteriormente. En el examen físico a los 2 años y 2 meses destacan peso, talla y PC bajo el P3, aspecto desnutrido y falta de panículo adiposo. Hipotonía generalizada, no se mantiene en sedestación y el sostén cefálico es incompleto. Lenguaje ausente, respiración ruidosa. La facies es triangular, con mordida invertida, leve prognatismo, orejas grandes despegadas y simples. Le realizamos cariotipo de alta resolución que muestra 46,XX del(14)(q11.2;q13.1) de novo. Se presentan estos 2 casos por ser una alteración cromosómica relativamente rara, revisar la literatura médica y discutir las manifestaciones clínicas, posibles genes candidatos presentes en la región delecionada y destacar la importancia del cariotipo de alta resolución.
Trisomía parcial 13q14.3-q22 derivada de una inserción-duplicación13q paterna
I. López, E. Guillén-Navarro, J.A. Bafallíu y A. Puche
Hospital Universitario Virgen de la Arrixaca. El Palmar. Murcia. España.
Introducción: El cromosoma 13 es el ejemplo más llamativo de viabilidad para un desequilibrio autosómico importante. La mayoría de la trisomías parciales del cromosoma 13 son derivadas de translocaciones o inversiones familiares. Presentamos un caso de duplicación parcial del cromosoma 13 derivado de una inserción-duplicación paterna.
Caso clínico: Se trataba de un niño derivado a los 6 años, por problemas de comportamiento y retraso en el lenguaje. En el estudio citogenético se observó un cromosoma 13 con material extra de 13q21. El cariotipo del padre, del abuelo paterno y de sus 2 hermanos también reveló un cromosoma 13 alterado, pero con menos material extra. Los estudios de hibridación in situ fluorescente (FISH) con sondas de cromosoma 13 completo, mostraron que todo el material deriva del mismo cromosoma. En estos casos, el patrón de bandas sugiere una duplicación 13q21.2-q22 insertada en 13q14.3. El paciente sería un recombinante con duplicación 13q14.3-q22 directamente insertada en 13q22. Están en proceso estudios de citogenética molecular para confirmar la anomalía descrita. El paciente a los 9 años presenta macrosomía, rasgos dismórficos leves y retraso mental moderado. RM cerebral y electroencefalograma (EEG), normales. El padre y el abuelo presentan hipoacusia pendiente de tipificar, sin otros hallazgos.
Discusión: Las alteraciones de la eucromatina, en contraste con la heterocromatina, tienen consecuencias fenotípicas, aunque en los últimos años se han publicado duplicaciones y deleciones familiares asociadas a fenotipo normal. La duplicación 13q21.2-q22 parece no tener efecto fenotípico en los cuatro portadores de esta familia, a excepción de la sordera en el padre y el abuelo. Hasta la fecha, nuestro caso sería el primero descrito con esta anomalía cromosómica.
Caracterización del cromosoma 15 en anillo presente en 2 pacientes con gran retraso de crecimiento mediante hibridación genómica comparada
C. Hernando1,2, P. Grao2, M. Santos1, M.J. García3, L. De la Maza4, J. Egozcue1 y C. Fuster1
1Departamento de Biología Celular, Fisiología e Inmunología. Unidad de Biología. Facultad de Medicina. Universitat Autònoma de Barcelona. 2Departamento de Genética. CERBA Internacional. Sabadell. Barcelona. 3Unidad de Neonatología. Hospital de la Santa Creu i Sant Pau. Barcelona. 4Sección de Endocrinología y Nutrición. Hospital General Yagüe. Burgos. España.
Introducción: El síndrome de cromosoma 15 en anillo es una anomalía cromosómica poco frecuente en la población (1/27.000 nacidos vivos). En la mayoría de los casos, la formación del cromosoma en anillo conlleva una pérdida de material genético. La característica fenotípica más frecuente es el importante retraso de crecimiento, tanto prenatal como posnatal, acompañado de retraso mental en grado variable y leves rasgos dismórficos.
Objetivo: Valorar la posible pérdida de material cromosómico en 2 pacientes, no emparentados, con un cromosoma 15 en anillo mediante la técnica de hibridación genómica comparada (CGH) y relacionar dicha pérdida con su expresión fenotípica.
Material y métodos: Estudio citogenético convencional (bandas G) y aplicación de la técnica de CGH.
Resultados: El análisis citogenético convencional mostró en ambos pacientes la presencia de un cromosoma 15 en anillo, r(15)(p11q26), en todas las metafases analizadas. El cariotipo de los padres fue normal. La técnica de CGH reveló que el tamaño del fragmento 15qter perdido era distinto; siendo el 15q26.2-qter para el paciente 1 y sospechando una pequeña deleción terminal de 15q26 en el paciente 2.
Conclusión: La combinación de los datos obtenidos mediante bandas G y CGH ha permitido identificar la región cromosómica delecionada. Ambos pacientes, a pesar de perder fragmentos cromosómicos de diferente tamaño, presentan retraso de crecimiento importante, sin embargo el paciente 2 (con menor pérdida de material cromosómico) presenta además retraso psicomotor e intelectual, mientras el paciente 1 presenta un desarrollo neurológico normal, por tanto y de acuerdo con lo descrito en la literatura médica, no siempre existe una correlación entre la cantidad de material delecionado y la intensidad del fenotipo observado.
Tetrasomía 15q11-q13: caracterización de un cromosoma marcador supernumerario CMS (15) en un paciente con autismo
M. Salido1, B. Gener2, R. Flores2, O. Villa1, M. del Campo2, J.A. Muñoz3, F. Solé1 y L.A. Pérez Jurado2
1Laboratorio de Citogenética y Biología Molecular. Servicio de Patología. Hospital del Mar-IMAS. Barcelona. 2Unidad de Genética. Universitat Pompeu Fabra. 3Servicio de Neuropediatría. Hospital del Mar-IMAS. Barcelona. España.
Presentamos un varón de 15 años con fenotipo autista según los criterios DSM-IV. Tras una historia prenatal y perinatal irrelevante el paciente presentó un retraso global del desarrollo motriz y del lenguaje con balbuceo inicial y bisílabos aunque sin progresión posterior del habla, así como epilepsia desde los 3 años con un foco irritativo frontotemporal izquierdo en el EEG. Fue intervenido a los 14 años por escoliosis. Actualmente presenta un trastorno claro de socialización asociado a retraso mental profundo. Su crecimiento se encuentra en el P10 con microcefalia límite (2 DE) y no presenta rasgos dismórficos significativos. Los potenciales evocados auditivos y visuales, y los estudios de imagen cerebral (TC y RM) han sido normales. El cariotipo mostró un cromosoma supernumerario marcador (47, XY, + mar [15]) que caracterizado por hibridación in situ fluorescente (FISH) con la sonda LSI Prader-Willi/Angelman, se corresponde con un inv dup(15) dicéntrico en ambos extremos. Los resultados de FISH indican que el cromosoma marcador contiene dos copias adicionales del gen SNRPN (15q11-q13), resultando en una tetrasomía de esta región. La reacción en cadena de la polimerasa (PCR) específica de metilación en SNRPN mostró una relación cromosoma materno frente a paterno de 3:1. Mediante microsatélites se han determinado los límites del fragmento tetrasómico y se ha confirmado el origen materno del cromosoma marcador, que incluye completamente la región delecionada en los SPW/SA, con un punto de rotura y unión que se corresponde con las duplicaciones segmentarias distales de dicha región. Inv dup(15) es el cromosoma marcador supernumerario más frecuente en seres humanos, aunque en la mayoría de los casos no se asocia a fenotipo posiblemente porque los reordenamientos ocurren en los puntos de rotura proximales a la región PWACR (región crítica del síndrome Prader-Willi/Angelman), delecionada en estos síndromes. En nuestro caso, en otros casos de inv dup y en las duplicaciones recíprocas de la región SPW/SA, probablemente el exceso de dosis génica de UBE3A (ubiquitín-proteína ligasa) ± otros genes (¿GABR B3, G3, A5?) sean responsables del fenotipo autista. Dado que parece que las anomalías citogenéticas o submicroscópicas de la región 15q11-q13 de origen materno son relativamente prevalentes en pacientes con autismo, debería considerarse su estudio detallado en todos los casos.
Agradecimientos: Trabajo subvencionado en parte por Fundació Parc de Recerca Biomédica de Barcelona, Red de centros de Genética (G03/07). Red de centros de Cáncer del fondo de investigación sanitaria ref. C03/07.
Síndrome de Morris y deficiencia mental ¿otro caso de síndrome de genes contiguos en Xq11-12?
F.J. Ramos1, I. Bueno1, M. Bassecourt2, M.J. López-Moreno1, J.L. Olivares1 y N. Dahl3
1Departamento de Pediatría. Facultad de Medicina. Hospital Clínico Universitario. Universidad de Zaragoza. 2Laboratorio de Citogenética. Hospital Infantil Miguel Servet. Zaragoza. España. 3Unit Clinical Genetics. Department of Genetics and Pathology. Uppsala University Hospital. Suecia.
El síndrome de Morris o síndrome de insensibilidad completa a los andrógenos (SICA) se produce por un defecto del receptor androgénico en la región Xq11-12. Las pacientes presentan un fenotipo con genitales externos femeninos pero con una fórmula cromosómica masculina (46,XY). Presentamos el caso de una niña con rasgos dismórficos y retraso psicomotor grave cuyo cariotipo fue 46,XY, confirmado por hibridación in situ fluorescente (FISH). En la RM cerebral se encontraron defectos de mielinización en la zona supratentorial y una hipoplasia del cuerpo calloso. La presencia de hallazgos no habituales en pacientes con una determinada patología de origen genético sugiere la afectación de otros genes, generalmente cercanos al gen responsable del cuadro principal (síndrome de genes contiguos). Uno de los genes situados en la proximidad del receptor androgénico es el denominado OLP (oligophrenin-1) que codifica una proteína activadora Rho-GTPasa y del que se han encontrado mutaciones en pacientes con retraso mental inespecífico. En 1999 se describieron 2 hermanas con SICA, hipotonía, epilepsia, ataxia y malformaciones del SNC. Ambas tenían cariotipos masculinos. Se discuten las similitudes de nuestra paciente con los casos descritos en la literatura médica. El estudio molecular del gen OLP está pendiente de finalización.
Trisomía 4p por translocación balanceada materna
I. Bueno Martínez1, F. Ramos Fuentes1 y E. Galán2
1Genética-Pediatría. Hospital Clínico Universitario. Facultad de Medicina. Zaragoza. 2Citogenética. Hospital Materno-Infantil. Badajoz. España.
Introducción: La trisomía 4p es una anomalía cromosómica individualizada en 1970 por Wilson et al. Actualmente se conocen unos 50 casos publicados en la literatura médica, y la mitad de ellos se deben a una translocación balanceada en uno de sus progenitores.
Caso clínico: Presentamos una niña de 7 años con retraso mental y de crecimiento, dismorfia facial y anomalías esqueléticas y oftalmológicas, que presenta esta anomalía cromosómica por translocación materna. Segunda hija de padres sanos, no consanguíneos. Embarazo controlado de 39 semanas de gestación. Parto espontáneo, vaginal. No precisa reanimación. Peso 3.140 g; longitud 45 cm; perímetro craneal (PC), 33 cm. Pies planos valgos con retropié derecho. Período neonatal normal. Retraso psicomotor: deambulación a los 2 años y lenguaje a los 4 años. Enuresis. A los 9 meses fue intervenida del pie derecho. A los 2 años fue ingresada por neumonía. Lleva tratamiento ortopédico por cifoescoliosis y sigue educación especial.
Exploración: a los 7 años y 5 meses, peso 14,7 kg (< 3 %), talla 114 cm (< 3 %) y PC 47 cm (< 2 DE). Microsomía con microcefalia, cara alargada y triangular con frontal prominente y sinofria. Coloboma iridiano derecho. Alas nasales antevertidas, filtro largo poco marcado, paladar ojival, incisivos grandes y separados. Orejas de baja implantación con grueso hélix. Cuello corto, cifoescoliosis. Rigidez articular generalizada con pies planos valgos y braquidactilia. Resto normal. Pruebas complementarias: cariotipo de lactante: 46,XX normal; cociente intelectual: 58; ORL, normal; oftalmología: coloboma iridiano completo derecho; radiografía: cifoescoliosis; EO = EC - EEG, TC cerebral, y PEA normales. Cariotipo de alta resolución: 46,XX,der(13)t(4;13) (p12;p11). Padre: 46,XY, normal. Hermana: 46,XX, normal. Madre: 46,XX,t(4;13)(p12;p13). Se revisa y comenta la bibliografía.
Estudio citogenético de pacientes con hipospadias como única malformación congénita
O. Vall1, González Ribero1, M. Santos2, C. Hernando2, V. Seidel1 y C. Fuster2
1Servicio de Pediatría. Hospital Universitario del Mar. Universidad Autónoma de Barcelona. 2Unidad de Biología. Facultad de Medicina. Universidad Autónoma de Barcelona. España.
Introducción: Las nuevas técnicas de citogenética molecular están contribuyendo a una mejor caracterización de asociaciones genotipo-fenotipo. Las hipospadias con frecuencia están asociadas a otras malformaciones congénitas, lo cual dificulta enormemente la determinación de gen o genes que, al alterarse, producen su aparición.
Objetivo: Realizar un estudio citogenético a una serie de niños con hipospadias, como única malformación congénita mayor, con el fin de detectar posibles alteraciones cromosómicas asociadas.
Material y métodos: Cultivo de linfocitos procedentes de sangre periférica y obtención de preparaciones cromosómicas. Análisis cromosómico a partir de bandas G y de la aplicación de la técnica de hibridación in situ fluorescente multicolor (M-FISH) (24 colores).
Resultados: Se ha realizado el estudio citogenético a 21 niños con hipospadias. El análisis del cariotipo ha revelado en un paciente la existencia de una translocación robertsoniana, t(13;14), de novo y en otro una inversión polimórfica, inv(9)(p11q12). En el resto de pacientes, los cariotipos han sido normales. La M-FISH no ha detectado ninguna alteración cromosómica compleja o críptica que hubiera podido pasar desapercibida con la técnicas de citogenética clásica.
Conclusión: Hasta la fecha en este tipo de pacientes se han publicado muy pocas deleciones cromosómicas implicando a diferentes regiones (1q42-q44, 4p16.13, 7q34 y 11p13) y sólo 2 casos, no emparentados, que presentaban la misma translocación "aparentemente" equilibrada t(1;16)(q12;q12). Nuestro estudio, por el momento, no ha aportado ningún dato relevante, debido al bajo número de pacientes estudiado o probablemente a las técnicas empleadas. En estos momentos estamos pendientes de la aplicación de sondas subteloméricas específicas para detectar desequilibrios en estas regiones tan ricas en genes.
Agradecimientos: Financiación recibida por SAF (2003-03894) y CIRIT (2001, SGR-00201).
Identificación mediante técnicas de citogenética molecular del mosaicismo presente en un niño con retraso psicomotor y craneosinostosis
B. Gener1, M. Santos2, C. Hernando2, M.P. Botella3 y C. Fuster2
1Dismorfología y Genética Clínica. Hospital de Cruces. Baracaldo. Vizcaya. 2Unidad de Biología. Facultad de Medicina. Universidad Autónoma de Barcelona. 3Departamento de Pediatría. Hospital de Txagorritxu. Vitoria. España.
Introducción: Las técnicas de citogenética molecular, especialmente la hibridación genómica comparada (CGH), son de suma importancia en el diagnóstico clínico al identificar de forma rápida las ganancias o pérdidas de material genético presentes en una alteración cromosómica desequilibrada.
Objetivo: La identificación mediante CGH e hibridación in situ fluorescente multicolor (M-FISH) (24 colores) de la trisomía parcial en mosaico (46,XY/47,XY + mar), detectada previamente mediante bandas G, en un niño que presenta retraso psicomotor, craneosinostosis parcial de la sutura sagital, asimetría facial y camptodactilia bilateral.
Material y métodos: Cultivo de linfocitos y extracción de ADN a partir de sangre periférica. Aplicación de las técnicas de CGH y M-FISH.
Conclusión: La combinación de las técnicas de bandas G, M-FISH y CGH ha permitido la caracterización del cariotipo de este paciente. Las manifestaciones clínicas de este paciente son superponibles a las descritas en otros casos de síndrome de trisomía 15q25-qter. La variabilidad en la gravedad del fenotipo puede ser debida al grado y la distribución de mosaicismo. La confirmación diagnóstica en este paciente permitirá comprender con mayor precisión el síndrome y ofrecer un asesoramiento médico más adecuado.
Agradecimientos: Financiación recibida por SAF (2003-03894) y CIRIT (2001,SGR-00201).
Detección de una deleción cromosómica críptica en 1p22.3 mediante HR-CGH en una niña con marcado retraso y dismorfias portadora de una translocación "aparentemente" equilibrada
G. Rodríguez-Criado1, M. Santos2, C. Hernando2 y C. Fuster2
1Unidad de Dismorfología. Hospital Virgen del Rocío. Sevilla. 2Unidad de Biología. Facultad de Medicina. Universidad Autónoma de Barcelona. España.
Introducción: La técnica de hibridación genómica comparada, en especial, de alta resolución (HR-CGH) permite detectar de forma rápida pequeños desequilibrios del genoma (menores de 3 Mb), revelándose como una metodología muy útil en el diagnóstico clínico.
Objetivo: Identificar mediante HR-CGH posibles desequilibrios crípticos en una niña con marcado retraso psicomotor y dismorfias portadora de una translocación t(1;14)(p22.2;p11.2) "aparentemente" equilibrada.
Material y métodos: Cultivo de linfocitos y extracción de ADN a partir de sangre periférica. Aplicación de las técnicas de hibridación in situ fluorescente multicolor (M-FISH) y de HR-CGH.
Resultados: Los perfiles de HR-CGH ponen de manifiesto de una forma clara la pérdida de una pequeña banda cromosómica correspondiente a 1p22.3.
Discusión: Las deleciones del brazo corto del cromosoma 1 son clínicamente muy raras y la mayoría se asocian, como en nuestra paciente, a numerosas anomalías y retraso de crecimiento. Hasta este momento, es la deleción intersticial en 1p más pequeña que se ha descrito a nivel constitucional.
Agradecimientos: Financiación recibida por SAF (2003-03894) y CIRIT (2001, SGR-00201).
Telegenética: un portal de atención a consultas médicas de genética entre profesionales de la salud
M. Del Campo1, J. Del Valle1, M. Martí2, J.C. Cabrera2, L. Toledo2 y L.A. Pérez Jurado1
1Unidad de Genética. Universitat Pompeu Fabra. Barcelona. 2Hospital Materno-Infantil. Las Palmas de Gran Canaria. España.
El portal www.telegenética.org ha estado en funcionamiento desde febrero de 2003. Durante este tiempo se han realizado un número considerable de consultas, la mayoría desde el hospital materno-infantil de Las Palmas dentro de un proyecto piloto para la validación metodológica y de utilidad del portal. En estas consultas se ha realizado un intento diagnóstico de cada caso y se han transmitido de manera detallada las recomendaciones médicas, de apoyo social y educativas propias de cada caso, así como el consejo genético correspondiente. El número y tipo de consultas hasta la actualidad (total, 71), así como el resultado de estas, ha sido el siguiente: dismorfología, 49; diagnóstico prenatal, 6; enfermedad genética, 5, y consulta puntual y se ha alcanzado un diagnóstico preciso en 56. El portal ha recibido 3.368 visitas. El 86 % proceden de España, México (2,6 %), Estados Unidos (1,7 %), Perú (1,5 %) y Argentina (1,4 %). Como se propuso, se ha iniciado en telegenética un foro de discusión para expertos integrantes de la red, abierto a otros profesionales. En este foro se han colgado los casos para los que no hay todavía diagnóstico. Los médicos usuarios han expresado su satisfacción con el portal como herramienta de apoyo diagnóstico.
Orphanet: un servicio europeo de información sobre enfermedades raras
P. Sancho, M. Del Campo y L.A. Pérez Jurado
Unidad de Genética. Universitat Pompeu Fabra. Barcelona. España.
Las llamadas enfermedades raras son enfermedades que afectan a una persona por cada 2.000 o menos, por lo que existirían en España alrededor de 20.000 pacientes con estas enfermedades. Orphanet es un servidor de información sobre enfermedades raras y medicamentos huérfanos de libre acceso para uso público, creado en Francia en 1996. Desde 2000, fondos de la comisión europea han facilitado la extensión de la recogida de datos a otros 7 países europeos, incluida España. La recogida de datos españoles se elaboró a partir de directorios ligados al área y búsquedas por internet. Se contactó por correo ordinario y/o electrónico a 279 asociaciones de pacientes, 60 laboratorios de diagnóstico, 60 consultas especializadas y 154 proyectos de investigación, con una respuesta del 24,37 %, 51,67 %, 41,67 % y 20,78 %, respectivamente. Orphanet ofrece servicios adaptados a las necesidades de los enfermos y sus familias, profesionales de la salud e investigadores, asociaciones de pacientes e industria. Orphanet facilita la investigación y la formación en esta área e incrementa el acceso a la información sobre enfermedades raras a pacientes y profesionales de la salud. Por ello, es necesaria la colaboración de las asociaciones de pacientes, especialistas e investigadores en este campo.
Estudios moleculares en pacientes con síndromes de sobrecrecimiento. Informe preliminar
P. Lapunzina1, A. Delicado1, L. Magano2, D. Arjona3, M.A. Mori1, C. Amiñoso3, M.L. de Torres1, L. Fernández1, M.C. Roche4, M. Palomares1, I. Incera2, P. Arias 2, R. Gracia5 e I. López Pajares1
Servicios de 1Genética Médica, 2Genética Molecular, 3Secuenciación Automática, 4Neurología Infantil y 5Endocrinología Infantil. Hospital Universitario La Paz. Madrid. España.
Introducción: Los síndromes de sobrecrecimiento (SSC) son un grupo heterogéneo de enfermedades que se caracterizan por presentar al menos dos de los tres parámetros antropométricos clásicos (peso, talla, perímetro cefálico) por encima del percentil 95 o +2 DE por encima de la media para edad y sexo. Se caracterizan además por presentar mayor riesgo de desarrollar retraso mental y una frecuencia mayor de tumores en la edad infantil o la adolescencia temprana que en la población general.
Pacientes y métodos: El Registro de Síndromes de Sobrecrecimiento recientemente iniciado en el Hospital Universitario La Paz contabiliza actualmente 112 individuos con síndrome de sobrecrecimiento. Se han realizado estudios moleculares de los genes CDKN1C, NSD1, GPC3, marcadores microsatélites polimórficos de la región 11p, 5q35 y Xq26 y Southern blot para diagnóstico de deleciones del cromosoma X, para aquellos pacientes con diagnóstico clínico sugerente de síndromes de Beckwith-Wiedemann, Sotos y Simpson-Golabi-Behmel. En los pacientes con diagnóstico de síndrome de Beckwith-Wiedemann se han realizado estudios de hibridación in situ fluorescente (FISH) con sonda específica de la región 11p.
Resultados: Se han hallado alteraciones en las tres enfermedades estudiadas. En pacientes con síndrome de Beckwith-Wiedemann se ha hallado disomía uniparental, duplicación 11p15.5 derivado de translocación paterna balanceada y mutación puntual del gen CDKN1C. En pacientes con síndrome de Sotos: deleción de la región 5q35 y mutaciones puntuales del gen NSD1 (inserciones, deleciones y mutaciones puntuales con y sin sentido), y deleción exónica y mutación puntual en el gen GPC3 en varones con síndrome de Simpson-Golabi-Behmel.
Conclusiones: Los estudios moleculares en síndrome de sobrecrecimiento confirman el diagnóstico clínico presuntivo y son de utilidad para el seguimiento de los pacientes y el asesoramiento de la familia.
Gigantismo de origen prenatal con mutación en el genNSD1, con evolución inicial sugerente de síndrome de Marshall-Smith
F.J. Ramos1, I. Bueno1, P. Ventura1, J. Del Valle2, M. del Campo2, J.L. Olivares1, L.A. Pérez-Jurado2 y J. Pérez-González1
1Sección de Genética. Departamento de Pediatría. Facultad de Medicina. Hospital Clínico Universitario. Universidad de Zaragoza. 2Unidad de Genética. Universidad Pompeu Fabra. Barcelona. España.
Presentamos un varón de 3 años con hipercrecimiento prenatal y posnatal inicialmente diagnosticado de síndrome de Sotos, cuya evolución tórpida los primeros meses de vida hizo pensar en un síndrome de Marshall-Smith.
Embarazo de 38 semanas con amenaza de parto prematuro y alfafetoproteína elevada, cariotipo prenatal 46,XY. Recién nacido con peso de 5.070 g y 58 cm de estatura, con un perímetro craneal (PC) de 39 cm. Test de Apgar 8/9. Fractura de clavícula. Hipoglucemia e hipercalcemia neonatal. Historia familiar sin interés para el caso. En la exploración física (4 meses) destacaba: peso, 7,600 kg (75-80 %); longitud, 70 cm (> 98 %); PC, 45,5 cm (> 97 %). Dolicocefalia, frente amplia, hipertelorismo ocular, filtro alargado, paladar elevado, dos incisivos inferiores (inició a los 3 meses), micrognatia y orejas grandes. Pequeña hernia umbilical. Hipotonía axial. Llanto ronco y ronquidos nocturnos. Exploraciones complementarias: Edad ósea: 9 meses; radiografía de manos, leve ensanchamiento de falanges medias; RM cerebral, ventriculomegalia leve, sin anomalías estructurales cerebrales. Evolución: A partir del quinto mes comenzó con pérdida de peso progresiva y enlentecimiento de velocidad de crecimiento, sin enfermedad aguda concomitante ni problemas de alimentación. Por este motivo precisó varios ingresos hospitalarios, algunos por enfermedad pulmonar aguda, hasta los 10 meses de vida. La edad ósea permanecía acelerada más de 2 años y persistía el retraso psicomotor. En este momento se consideró que podría tratarse de un síndrome de Marshall-Smith. Sin embargo, a partir del mes 11 el paciente recuperó su ritmo de crecimiento pondoestatural, con talla entre los percentiles 90-97, que se ha mantenido hasta la fecha. No hubo más episodios de infecciones respiratorias graves y persistía el retraso psicomotor, especialmente en el área del lenguaje y motórica gruesa. La evolución de este paciente hacía necesario aclarar el diagnóstico definitivo, por lo que se realizó el estudio molecular del gen NSD, implicado en el síndrome de Sotos. El análisis del dicho gen, localizado en 5q35, detectó una mutación de novo heterozigota consistente en una deleción de 10nt en el exón 7 que provoca un cambio de lectura y da lugar a un codón de stop precoz. Este caso ilustra la posibilidad de solapamiento fenotípico y evolutivo entre el síndrome de Sotos y el síndrome de Marshall-Smith y justificaría la realización del análisis mutacional del gen NSD1 en pacientes con este último síndrome.
Consecuencias fenotípicas de la mutación r625w del gengli3en una familia
E. Guillén Navarro1, J.L. Alcaraz León2, E. Martínez Villalta1 y M. Kalff-Suske3
1Unidad de Genética Médica y Dismorfología. 2Sección de Neonatología. Servicio de Pediatría. Hospital Universitario Virgen de la Arrixaca. Murcia. España. 3Zentrum fuer Humangenetik. Philipps-Universitaet Marburg. Alemania.
Introducción: Se ha identificado haploinsuficiencia del gen GLI3 en varios síndromes malformativos que comparten anomalías digitales (morfopatías GLI3): síndrome de cefalopolisindactilia de Greig (MIM 175700), síndrome de Pallister-Hall (MIM146510), polidactilia preaxial tipo IV (MIM 174700), polidactilia postaxial tipo A (MIM 1472000) y polidactilia postaxial tipo A/B. A. Diversos autores han sugerido cierta correlación genotipo-fenotipo. Se presenta la variabilidad fenotípica intrafamiliar del síndrome de cefalopolisindactilia de Greig (SCPSG) como consecuencia de la mutación R625W en el gen GLI3.
Casos clínicos: La paciente es una recién nacida, hija de padres jóvenes y no consanguíneos, con frente prominente, raíz nasal ancha, hipertelorismo, microrretrognatia leve, hernia supraumbilical, sindactilia cutánea parcial de segundo y tercer dedos y total de tercer, cuarto y quinto dedos de ambas manos y polidactilia preaxial con sindactilia de los restantes dedos de ambos pies. Su madre presentaba prominencia frontal, sindactilia cutánea total intervenida del tercer y cuarto dedos de mano izquierda, sindactilia cutánea parcial del segundo y tercer dedos de ambas manos, polidactilia preaxial bilateral en pies intervenida, sindactilia cutánea total del segundo y tercer dedos y parcial del tercer y cuarto dedos de pies. La abuela materna fallecida era el único caso con polidactilia postaxial mínima. La bisabuela presentaba polidactilia preaxial y sindactilia de pies y la tatarabuela sólo sindactilia de pies. Se refieren las manifestaciones fenotípicas de otros miembros de la familia. Estudio citogenético en la niña y su madre demuestra un cariotipo normal. El estudio molecular del gen GLI3 en ambas pacientes demuestra un cambio c.1873C > T (R625W).
Comentarios: 1. Esta familia confirma la expresividad variable del SCPSG. En la mayoría de casos la exploración clínica detallada nos lleva al diagnóstico. 2. La mutación R625W en el gen GLI3 es responsable del fenotipo observado. El estudio molecular del gen GLI3 puede ser útil en identificar casos con manifestaciones muy leves. 3. Hasta la fecha se han descrito 24 mutaciones puntuales diferentes en GLI3 asociadas a SCPSG. El R625W se ha identificado previamente en otra familia de cuatro generaciones, donde fue transmitida por una madre asintomática a sus dos hijos afectados clínicamente, lo que sugiere una penetrancia incompleta que contradice suposiciones previas. 4. La posible penetrancia incompleta en el SCPSG es de especial interés a la hora de indicar e interpretar los resultados de los estudios moleculares.
Síndrome de Smith-Lemli-Opitz
T. Vendrell1, J. de la Torre2, M. Girós1, A. Mas2, E. Carreras3, J. García3 y L. Torán2
1Unidad de Genética. 2Servicio de Anatomía Patológica. 3Unidad de Ecografía Obstétrica. Hospital Vall d'Hebron. Barcelona. España.
El síndrome de Smith-Lemli-Opitz es un síndrome dismorfológico con un patrón de herencia autosómica recesiva y un amplio espectro de anomalías fenotípicas, de las que cabe destacar las características faciales, polisindactilia, anomalías genitales y malformaciones cardíacas y pulmonares. Está causado por la mutación del gen codificador de la enzima 7-deshidrocolesterol reductasa que transforma el 7 deshidrocolesterol en colesterol (DHCR-7). El diagnóstico de los pacientes se establece por el patrón dismórfico y se puede confirmar mediante análisis bioquímico que se basa en la determinación de concentraciones bajas de colesterol con incremento de 7-DHC. Esta determinación permite el diagnóstico diferencial frente a otros cuadros dismórficos y por tanto el consejo genético adecuado y la posibilidad de diagnóstico prenatal. Presentamos 2 casos de diagnóstico de síndrome de Smith-Lemli-Opitz en fetos de 22 y 21 semanas de gestación. En el primer caso el diagnóstico se sugirió por las malformaciones detectadas en el estudio ecográfico prenatal: polidactilia postaxial de ambas manos y pie derecho, cardiopatía y genitales ambiguos con cariotipo 46,XY. Este diagnóstico se confirmó con la determinación en líquido amniótico de valores elevados de 7-DHC y posteriormente fue posible la detección de la mutación responsable. Estos resultados han permitido el estudio prenatal en un posterior embarazo que corresponde a un feto no afectado. En el segundo caso se sugirió el diagnóstico a partir de la exploración y los datos del estudio autópsico. Se observó fenotipo dismórfico con micrognatia, orejas de implantación baja, puente nasal ancho y plano, polidactilia postaxial en pie derecho e hipospadias peneano asociados a otras malformaciones internas también propias de este síndrome, agenesia renal izquierda, arteria umbilical única y fallo de lobulación pulmonar. En este caso no fue posible el estudio bioquímico y está pendiente de estudio molecular. En una próxima gestación, además de un estudio ecográfico detallado, deberá obtenerse líquido amniótico para diagnóstico bioquímico.
Probable nuevo síndrome de RM/MAC ligado a X
G. Rodríguez Criado1, G. Meroni2, F. Yanes1 y I. Gómez de Terreros1
1Unidad de Dismorfología del Hospital Universitario Virgen del Rocío. Sevilla. España. 2TIGEM. Nápoles. Italia.
Objetivo: Diagnosticar a los componentes con retraso mental de cuatro generaciones de una familia.
Pacientes y método: Exploración clínica de los cinco miembros a los que hemos tenido acceso, y ubicación dentro del árbol genealógico familiar de los otros miembros fallecidos o no con retraso mental. Estudio comparativo de nuestros enfermos con las bases de datos Oxford Medical Database de Neurogenética y Dismorfología, POSSUM, así como otras bases de datos colocadas en internet y el amplio material bibliográfico del que disponemos. Estudios de ligamientos a partir de 13 muestras de sangre de la familia, 3 enfermos vistos por nosotros, uno posiblemente enfermo visto en otro centro y nueve parientes sanos. No se dispuso de los otros dos enfermos conocidos vivos por razones de imposibilidad física, por ahora. Estudio, en los 4 enfermos, del gen MID1 y ALPHA4, un gen interactor de MID1, por el parecido clínico de estos enfermos con el síndrome de Opitz.
Resultados: El árbol genealógico reveló una herencia ligada a X recesiva, con 9 individuos de 4 generaciones que probablemente padecen el mismo cuadro clínico, 6 vivos y 3 fallecidos. El cuadro clínico se caracteriza por retraso mental leve o moderado en el 100 % de los casos y los siguientes signos en un porcentaje variable: talla corta, microcefalia, cara pequeña, boca pequeña, pene pequeño y escroto hipoplásico, criptorquidismo, cardiopatía congénita. El resto pormenorizado de la sintomatología se expondrá en el Congreso. Las pruebas rutinarias, cariotipo de alta resolución, Fra X, AA en sangre y orina, ácido láctico y pirúvico, amoniemia fueron normales en los enfermos estudiados. No se encontraron mutaciones ni en MID1 ni en ALPHA4. El estudio de ligamiento ha mapeado al probable gen causante del RM de esta familia en Xp11.3-q21.32 con un LOD score de 2. Se está intentando estrechar la banda en el TIGEM de Nápoles por la Dra. Meroni en la actualidad y probablemente no esté concluido a primeros de marzo.
Conclusiones: Se describe lo que nosotros creemos se trata de un nuevo síndrome de retraso mental y múltiples anomalías congénitas, ligado al cromosoma X.
Síndrome de distrés respiratorio del recién nacido: diagnóstico molecular del déficit de Sp-B
M. González-Rivera1, J.L. Jiménez-Fuentes1, Y. López Lozano2, A. Arroyos Plana2, A. Martínez Colom3, M. Sánchez-Luna4 y M.A. Muñoz-Fernández3
1Línea Instrumental Secuenciación. Hospital General Universitario Gregorio Marañón. Madrid. 2Servicio de Neonatología. Complejo Hospitalario Toledo. Hospital Virgen de la Salud. 3Laboratorio de Inmunobiología Molecular. 4UCIN. Servicio de Neonatología. Hospital General Universitario Gregorio Marañón. Madrid. España.
Antecedentes: La enfermedad pulmonar aguda del neonato es la causa más frecuente de morbimortalidad en niños menores de un año. En prematuros, la causa más frecuente del síndrome de distrés respiratorio (SDR) es el déficit de surfactante, que revierte con la maduración pulmonar. En el neonato a término son múltiples las causas, y se han descrito dos grupos de marcadores genéticos, los ligados a la pérdida de la función surfactante (ausencia de la proteína B del surfactante pulmonar) y aquellos que estadísticamente están asociados al riesgo de padecer distrés respiratorio del recién nacido.
Caso clínico: Neonato a término, mujer, primer hijo de padres sanos, no consanguíneos, que inicia SDR desde las primeras horas de vida, requiriendo ventilación mecánica convencional. La radiografía de tórax presenta condensaciones bilaterales generalizadas, con alveolograma aéreo y broncograma. Precisa numerosas dosis de surfactante exógeno natural (Curosurf), mejorando, de forma transitoria, clínica y radiológicamente. La evolución del cuadro clínico desembocó en el fallecimiento del neonato a los 27 días de vida. No existieron antecedentes familiares de enfermedades respiratorias ni endogamia.
Objetivo: Investigar las mutaciones asociadas al déficit congénito de proteína B del surfactante pulmonar en el neonato afectado y en sus padres, además de documentar la historia clínica en relación con el estudio genético.
Material y métodos: Se realizó extracción de ADN (High Pure PCR Template Preparation Kit-Roche Diagnostics) desde sangre total, amplificación del gen SFTPB (AF 400074) con oligonucleótidos validados (Nogee LM, 2000) y secuenciación de 10 exones (BigDye Terminator v.3.1, Applied Biosystems) en un secuenciador ABI PRISM 3100. Las secuencias se alinearon frente a la consenso gi15021770).
Resultados: A los 25 días de vida se confirmó la sospecha del déficit congénito de Sp-B mediante estudio molecular, presentando el neonato afectado la mutación 121ins2 en homozigocia (exón 4) que consiste en la sustitución de la 1549C por el triplete GAA, lo que ocasiona un cambio en la fase de lectura originando un codón de parada prematura, con completa ausencia del precursor de la proteína B del surfactante pulmonar. Los padres presentaban esta mutación en heterozigocia. El estudio del resto de los exones no aportó ningún otro hallazgo mutacional. Este es el primer caso documentado de déficit congénito de Sp-B en España.
Isodisomía materna parcial en un caso de síndrome de Silver-Russell
J. Rosell1, M. Caimari2, A. Pérez1, L. Torres2, M. Bernués1 y D. Heine1
1Sección de Genética. 2Unidad de Endocrinología Infantil. Hospital Universitari Son Dureta. Palma de Mallorca. España.
El síndrome de Silver-Russell se caracteriza por presentar un retraso de crecimiento prenatal y posnatal, facies peculiar, clinodactilia, asimetría corporal y estar asociado en el 10 % de los casos a una disomía uniparental materna del cromosoma 7. Se presenta el caso de un niño de 2 años de edad remitido por el servicio de pediatría para valorar su talla baja. Se trataba del segundo hijo de una pareja sana de 44 y 45 años de edad, no consanguínea, con la gestación estudiada mediante amniocentesis con resultado normal de 46,XY, parto eutócico de 35 semanas con peso al nacimiento de: 1.950 g, talla 40,5 cm y perímetro craneal (PC), 32. La somatometría evolutiva siempre fue por debajo del tercer percentil. En la exploración física destacaba la existencia de la talla baja con facies característica sin otras manifestaciones clínicas. Se solicitaron los siguientes exámenes complementarios: seriada esquelética, cariotipo y estudio molecular para descartar la existencia de disomía uniparental materna. Los resultados obtenidos en el estudio radiológico se descarta la existencia de asimetría corporal, poniéndose de manifiesto un retraso en la maduración ósea y el cariotipo mostró una fórmula cromosómica normal de 46,XY. El estudio molecular indica que el paciente presenta una isodisomía materna parcial del cromosoma 7, lo que sería una forma poco habitual de presentación.
Seudohermafroditismo masculino, forma parcial de resistencia a los andrógenos, con mutación P378R en exón 1 del AR
N. Torán1, M. Fernández-Cancio2, L. Audí2, C. Piró1, M. Gusinyer3, J. De la Torre1 y A. Carrascosa3
1Servicio de Anatomía Patológica. Hospital Ramón y Cajal. Madrid. España. 2Unidad de Investigación de Endocrinología Pediátrica. 3Servicio de Endocrinología Infantil. Hospital Vall d'Hebron. Barcelona. España.
El proceso diagnóstico en los estados intersexuales debe comprender, actualmente, un completo estudio clínico, radiológico, hormonal, anatomopatológico y, finalmente, llegar, a ser posible, a la detección de la mutación responsable de la enfermedad. Se presenta el caso de un varón de origen gambiano, hijo de padres primos hermanos, con cariotipo 46,XY que en la ecografía morfológica a las 20 semanas de gestación presentaba genitales ambiguos. Nació a las 40 semanas, con un peso de 2.300 g, con talla de 45 cm. A los 5 días de vida se determinaron: cortisol, 14 m/dl; 17-OH-P, 15,2 ng/ml (N27 ± 18); 11 DOC 11,3 ng/ml (N6-38); D4 11 ng/dl (N35 ± 17); T 267 ng/dl (N33 ± 4). A los 3 meses: cortisol 6,2 mg/dl, 17-OH-P 2,2 ng/ml; a los 4 meses, 17-OH-P 58 ng/dl, D4 140 ng/dl, T 30 ng/dl. Clínicamente se detecta talasemia menor y asma bronquial. A los 9 meses inicia tratamiento con testosterona intramuscular durante 4 meses. A los 2 años presenta pene pequeño, hipospadias con meato en raíz del pene. Bolsas escrotales atróficas y vacías, escroto bífido. Gónadas no palpables. Uretra masculina. La ecografía pélvica evidencia estructura tubular que podría corresponder a útero y que se continúa con vagina. No se identifican gónadas. A los 3 años se practica enderezamiento del pene. Cistoscopia: estenosis del meato y orificio de desembocadura de la vagina. A los 4 años es sometido a laparoscopia: gónadas bilaterales de tamaño pequeño junto a restos de útero y trompas. Laparotomía: se extirpan gónadas de aspecto disgenético con pedículo vascular de unos 2 cm que sale del cuerno uterino. El estudio histológico identificó dos gónadas masculinas con parénquima testicular constituido por túbulos embrionarios con aplasia de células germinales. La albugínea muestra túbulos en su espesor. Los tejidos acompañantes comprenden: hidátide de Morgagni, epidídimo, deferente, rudimento de trompa de Falopio y útero hipoplásico. La morfología es consistente con seudohermafroditismo masculino con restos müllerianos. Posteriormente se detecta la mutación P378R en el exón 1 del gen AR. Diagnóstico final: seudohermafroditismo masculino por resistencia a los andrógenos en su forma incompleta.
Fenotipo acondroplasia con mutación de hipocondroplasia en 2 pacientes. Importancia del diagnóstico molecular en las osteocondrodisplasias
L.F. Magano1, P. Lapunzina1, I. Arroyo Carrera2, I. González Casado1, I. Incera1 y R. Gracia Bouthelier1
1Hospital Universitario La Paz. Madrid. 2Hospital San Pedro Alcántara. Cáceres. España.
Las osteocondrodisplasias son un grupo heterogéneo de enfermedades de causa genética que comprometen principalmente las estructuras óseas y articulares de los huesos largos y planos. Hasta hace unos pocos años, el diagnóstico de acondroplasia era meramente clínico, diagnóstico que usualmente no ofrece dificultades para el pediatra experimentado. Desde el descubrimiento del gen que afecta a esta enfermedad (FGFR3) es posible su estudio molecular, pudiéndose confirmar el diagnóstico en el 100 % de las oportunidades. Se presentan 2 pacientes que habían sido diagnosticados clínicamente de acondroplasia, cuyo estudio molecular reveló que estaban afectados de la mutación clásica de hipocondroplasia. Caso 1. GRR. Niño de 10 meses de edad en el momento de la consulta inicial. Nació de parto por cesárea por presentación de nalgas. Peso de nacimiento de 3.770 g; talla, 49 cm; perímetro craneal (PC), 39 cm. No requiere reanimación en el período neonatal. Establecen diagnóstico de acondroplasia en el lugar de nacimiento. Padres sanos, no consanguíneos; madre de 36 años; padre de 39. Ambos con talla alta-normal. Desarrollo psicomotor algo lento en los primeros 8 meses de vida, requiriendo rehabilitación psicomotora. Al examen físico a los 10 meses presenta peso de 8.700 g (P25-50); talla, 66,5 cm (3,8 DE); PC, 52 cm (> P97). Macrocefalia, raíz nasal hundida, puente nasal ancho con nariz corta, philtrum algo largo, extremidades cortas. Dedos de manos y pies cortos y toscos, con manos en seudotridente. Cifosis lumbar marcada. Radiografía de esqueleto compatible con el diagnóstico de acondroplasia. Estudio molecular para acondroplasia negativo. El estudio molecular para hipocondroplasia reveló una mutación N540K (C1620 * G). Caso 2. Niña de 14 meses en el momento de la consulta inicial en nuestro servicio. Nació de parto normal a las 34 semanas por amniorrexis. En las ecografías prenatales notaron polihidramnios, miembros cortos y macrocefalia. Peso de nacimiento de 2.700 g (75-90); talla, 45 cm (25); PC, 35 cm (> 90). Requiere reanimación superficial en el período neonatal. Sugieren diagnóstico de acondroplasia probable en el lugar de nacimiento. Padres sanos, no consanguíneos, madre de 37 años y padre de 37. Ambos con talla normal-media. Desarrollo psicomotor normal en su primer año de vida. Al examen físico a los 14 meses presenta peso 9.200 g (P25-50); talla, 68 cm (3,3 DE); PC, 50,5 cm (P97). Macrocefalia, raíz nasal hundida, puente nasal ancho con nariz corta y extremidades cortas. Dedos de manos y pies cortos y toscos, con manos en tridente. Hiperlordosis lumbar marcada. Radiografía de esqueleto compatible con el diagnóstico de acondroplasia. Estudio molecular para acondroplasia negativo. El estudio molecular para hipocondroplasia mostró mutación N540K (C1620 → A). Hasta la actualidad hemos estudiado casi un centenar de pacientes con mutaciones específicas de acondroplasia e hipocondroplasia, coincidiendo en general el fenotipo con el genotipo. Sin embargo, en estos 2 pacientes las características clínicas y radiológicas sugerían acondroplasia y el estudio molecular demostró hipocondroplasia. La importancia de un diagnóstico clínico y su confirmación molecular radica en un correcto consejo genético para el paciente y su familia, así como un asesoramiento sobre la evolución de talla y el desarrollo general del mismo, por lo que queremos destacar la importancia del estudio molecular en todos los pacientes con diagnóstico de acondroplasia o hipocondroplasia.
Síndrome de Kearns-Sayre debido a una deleción de 8,4 kb en el ADNmt
F.J. Ramos1, I. Bueno1, A. Jiménez1, E. López2, A. Solayo2, J.L. Olivares1, J. Montoya2 y M. Bueno1
1Departamento de Pediatría. Facultad de Medicina. Hospital Clínico Universitario. Universidad de Zaragoza. 2Departamento de Bioquímica y Biología Molecular. Facultad Veterinaria. Universidad de Zaragoza. España.
El síndrome de Kearns-Sayre (SKS) es una enfermedad neuromuscular que se caracteriza por: a) inicio en la infancia o adolescencia; b) oftalmoplejía externa crónica y progresiva, y c) degeneración retiniana. Otros hallazgos frecuentes son cardiopatía, hipoacusia, ataxia o alteraciones endocrinas (diabetes). La enfermedad se debe a deleciones del ADN mitocondrial que varían de 1 a 8 kb, sin que su tamaño parezca influir en el fenotipo clínico. Se mantiene que es el porcentaje relativo de mitocondrias mutadas el que determinaría el grado de afectación del paciente. Se presenta una niña de 16 años que a los 6 años presentó hipoacusia, ptosis palpebral bilateral e hipotonía muscular, manifestaciones que se han ido agravando con la edad, y han precisado el uso de audífonos y una escolarización con apoyos. La paciente presenta disfunciones neurológicas transitorias que en una ocasión dieron lugar a un deterioro progresivo de la conciencia con entrada en coma, que pudo superar. La paciente presenta un estado de malnutrición crónica y graves problemas de visión. Los estudios complementarios realizados mostraron una elevación del lactato/piruvato y la creatinfosfocinasa en sangre, hiperproteinorraquia y normalidad de AAs en sangre y orina. Se solicitó una biopsia muscular para descartar una enfermedad mitocondrial, pero no pudo realizarse por negativa familiar o por el mal estado general de la paciente. Se descartaron otras enfermedades neuromusculares como miastenia grave, distrofia miotónica y distrofia facioescapulohumeral. Un examen oftalmológico demostró la presencia de lesiones pigmentarias en la retina y la RM cerebral mostró heterotopias de la sustancia blanca con anomalías del patrón de mielinización. Finalmente, se pudo realizar la biopsia muscular, y se demostró la presencia de una gran deleción (8,4 kb) en el ADN mitocondrial del 82 % de las mitocondrias estudiadas. Esta deleción es una de las de mayor tamaño descritas en la literatura médica.
Caracterización Molecular de la región 5q35 alrededor del gen NSD1
J.M. Del Valle, M. del Campo y L.A. Pérez Jurado
Unidad de Genética. Universitat Pompeu Fabra. Barcelona. España.
El síndrome de Sotos (SS) es un trastorno autosómico dominante caracterizado por un excesivo crecimiento prenatal y postnatal, edad ósea adelantada, rasgos craneofaciales característicos y diversos grados de retraso mental. Está originado por una haploinsuficiencia del gen NSD1, bien por deleciones que incluyen todo el gen o mutaciones puntuales hipo o amórficas de NSD1. Las deleciones submicroscópicas de 2,2 Mb son frecuentes en pacientes japoneses y representan dos terceras partes de los alelos mutados. Se ha estudiado la estructura de la región genómica 5q35 en controles y en pacientes con síndromes de Sotos, así como en algunos primates. La región presenta bloques de duplicaciones segmentarias (DS) de un tamaño de 143, 148 y 51 kb, respectivamente, y con una identidad de secuencia mayor o igual al 99 %, uno centromérico a NSD1 y dos teloméricos separados por 2,2 Mb y situados invertidos el centromérico y el primer telomérico, y en tándem el centromérico y el segundo telomérico. Existen 2 bloques de DS adicionales en 16p11.2. El estudio mediante microsatélites localizados en las DS sugiere la existencia de un número variable de DS entre individuos, al menos para las copias del cromosoma 16. El análisis de muestras de primates hominoides indica que estas DS han aparecido muy recientemente en la evolución genómica. En nuestra serie de 26 pacientes con diagnóstico clínico de síndrome de Sotos y uno con síndrome de Weaver, se ha descartado la existencia de deleciones en 5q35. El estudio mutacional por el momento ha revelado una mutación patogénica en un paciente y varios polimorfismos. La ausencia de deleciones submicroscópicas en nuestra serie, y el bajo porcentaje hallado en otros estudios en pacientes europeos respecto a los pacientes japoneses, sugiere que exista variabilidad en el número u orientación de las DS que suponga alelos de predisposición con frecuencias diferentes entre poblaciones.
Análisis mutacional y de las actividades de proteínas mutadas en pacientes españoles con la enfermedad de Gaucher
P. Alfonso1, J.C. Rodríguez-Rey2, A. Gañán1, J.I. Pérez-Calvo3, P. Giraldo4 y M. Pocoví1
1Departamento de Bioquímica y Biología Molecular y Celular. Facultad de Ciencias. Universidad de Zaragoza. 2Departamento de Biología Molecular. Facultad de Medicina. Universidad de Cantabria. Santander. 3Servicio de Medicina Interna. Hospital Clínico Universitario Lozano Blesa. Zaragoza. 4Servicio de Hematología. Hospital Universitario Miguel Servet. Zaragoza. España.
La enfermedad de Gaucher (EG) es una de las enfermedades hereditarias lisosomales más frecuentes, debida a un déficit en la enzima lisosomal glucocerebrosidasa. La EG presenta una gran variabilidad, tanto en la sintomatología como en la respuesta de los pacientes al tratamiento enzimático sustitutivo (TES). Esta heterogeneidad se atribuye en parte al gran número de mutaciones que se encuentran en el gen de la glucocerebrosidasa y a la actividad residual de las proteínas mutadas. Por ello, el objetivo principal de este trabajo fue identificar las mutaciones responsables de la enfermedad en un grupo de 51 pacientes españoles, caracterizarlas y conocer la actividad residual de las enzimas recombinantes portadoras de mutaciones nuevas, para poder así predecir la evolución clínica de los pacientes con la EG y su respuesta al TES. Para conseguir este objetivo de identificación y caracterización de las mutaciones, se emplearon técnicas basadas en la reacción en cadena de la polimerasa (PCR), conformación de polimorfismos de cadena sencilla (SSCP), secuenciación automática y análisis con endonucleasas de restricción. Mediante estas técnicas se identificaron un total de 27 alelos mutados diferentes, lo cual supuso la caracterización del 100 % de los alelos mutados. Para comprobar que las mutaciones nuevas encontradas eran causales de la EG, se introdujeron mediante mutagénesis dirigida los cambios correspondientes en el ADNc de la glucocerebrosidasa y se transfectaron en células COS-7. Las actividades enzimáticas de las proteínas expresadas fueron medidas y comparadas con la actividad de la glucocerebrosidasa silvestre humana, rindiendo actividades que oscilaban entre 5,5 y 43 % de la actividad considerada normal. Nuestros resultados demuestran que las mutaciones encontradas son las causales de la EG. Sin embargo, las actividades residuales de las proteínas portadoras de dichas mutaciones no son suficientes para explicar el fenotipo clínico de la EG.
Perfil de distribución epidemiológica y genética de la enfermedad de Gaucher en España
P. Giraldo1, M. Pocoví2, P. Alfonso2, J.I. Pérez-Calvo3 y M. Giralt1 por el Grupo español de enfermedad de Gaucher
1Hospital Universitario Miguel Servet. Zaragoza. 2Universidad de Zaragoza. 3Hospital Clínico Universitario. Fundación Española para el estudio y Terapéutica de la Enfermedad de Gaucher (FEETEG). España.
La EG es la más frecuente de las enfermedades por error congénito del metabolismo de los esfingolípidos, producida por déficit de actividad enzimática β -glucocerebrosidasa ácida debido a la alteración en la secuencia del gen que la codifica. El conocimiento del genotipo distingue formas neuronopáticas y no neuronopáticas; sin embargo, es poco preciso para predecir la gravedad de la enfermedad, especialmente en el tipo 1, pacientes con la misma alteración genética están asintomáticos y otros muy afectados. El objetivo del estudio fue analizar los factores genéticos y ambientales responsables de la heterogeneidad en la expresión clínica y evaluar las causas de la variabilidad interindividual en la respuesta al tratamiento enzimático sustitutivo (TES).
Pacientes y métodos: Desde 1993 se dispone de un registro informatizado de pacientes españoles afectados de EG, obtenido mediante encuesta, en el que hay recogidos datos demográficos/antropométricos, clínicos, biológicos, esqueléticos, genotipo y evolutivos. Así mismo se dispone de muestras de ADN genómico individuales de 200 pacientes y muestras de plasma obtenidas en el momento del diagnóstico enzimático realizado en nuestro laboratorio y muestras de plasma de 120 pacientes que están sometidos a TES. Se ha realizado análisis descriptivo, estudio de correlación genotipo/fenotipo y análisis multivariante para identificar factores relacionados con la enfermedad ósea.
Resultados: A 30 de junio de 2003 se había recibido información de 557 sujetos pertenecientes a 182 familias no relacionadas. Los pacientes incluidos en el REEG (262) procedían de 66 centros sanitarios. Se han identificado 242 pacientes tipo 1 y 18 tipo 2/3. En el 93 % de los pacientes se ha identificado la mutación responsable del defecto. El genotipo más frecuente era el N370S/L444P, los otros genotipos representaban el 64,0 % del total. Once de los genotipos se han descrito por primera vez en España. La correlación entre datos clínicos/biológicos y el genotipo muestra que los pacientes N370S/N370S tenían manifestaciones clínicas más leves, edad al diagnóstico superior (p < ,0002) e índice de gravedad significativamente menor (p < 0,0455) que los pacientes con otros genotipos. La enfermedad ósea como manifestación clínica más limitante para el paciente con EG tipo 1 no tiene relación aparente con el genotipo ni con la actividad enzimática residual. En el análisis multivariante las variables sexo femenino (p = 0,057), esplenectomía previa (p = 0,043) e índice pronóstico superior a 7 (p = 0,001) eran significativas para la presencia de enfermedad ósea.
Conclusiones: El abordaje multidisciplinar de la EG permite integrar conocimientos genéticos con la expresión clínica y evolutiva y disponer de un modelo aplicable a otras entidades.
Enfermedad de Chanarin-Dorfman presentándose como eritroqueratodermiavariabilis
M. Gilaberte1, A. Azón2, A. Toll1, A. González Enseñat3, S. Bel2, L. Florensa4, J. Lloreta5, J. Fischer6 y R.M. Pujol1
Servicios de Dermatología. 1Hospital del Mar. 2Sant Joan de Reus (Tarragona) y 3Hospital Sant Joan de Déu (Barcelona). Servicios de 4Hematología, 5Anatomía Patológica. Hospital del Mar de Barcelona. España. 6Centro National de Gènotypage Evry Cedex. Francia.
Introducción: El síndrome de Chanarin-Dorfman (SCD) es una rara enfermedad genética de transmisión autosómica recesiva. El gen del SCD, denominado NCIE2, se haya en el cromosoma 3p21. Se trata de una metabolopatía de depósito en la cual una alteración del catabolismo de los triglicéridos hace que estos se acumulen en diferentes órganos (piel, hígado, músculo, médula ósea, nervio, ojo, etc.), produciendo diversas manifestaciones. El SCD es muy heterogéneo tanto en la sintomatología como en la intensidad de su presentación. En la piel suele cursar como una ictiosis congénita o una eritrodermia ictiosiforme no ampollosa. Presentamos un caso de SCD con una forma de presentación no descrita: la eritrodermia variabilis (EQV).
Caso clínico: Niño de 4 años de edad, sin antecedentes familiares de interés, segundo hijo de madre sana con desarrollo psicoponderoestatural normal. Desde el nacimiento presentaba cuadro ictiosiforme, eritematodescamativo, que en tronco y extremidades formaba placas extensas de borde activo, bien delimitadas, simétricas y migratorias. Existía afectación facial, capilar y en mayor medida en codos y rodillas. La dentición, uñas y sudoración eran normales. Las exploraciones neurológica, ofatalmológica y ORL eran normales hasta la fecha. Exploraciones complementarias: Analítica básica: hipertransaminemia persistente, elevación de enzimas musculares. Aminoacidemia: valores bajos de treonina, isoleucina y asparragina. Inmunoglobulinas, cobre, cinc, ceruloplasmina normales. Biotinidasa: disminuida (estado de portador). Biopsia cutánea: hiperqueratosis con paraqueratosis. Estudio del cabello: normal. Seriada ósea: normal. Extensión de sangre periférica: presencia de múltiples vacuolas en leucocitos, neutrófilos y linfocitos. ME de leucocitos en sangre y piel: vacuolas lipídicas intracitoplasmáticas. Estudio genético: gen NCIE2 con mutación puntual en el exón 6 (mutación sin sentido): transmitida por el padre y mutación (inserción/deleción) en el exón 4 transmitida por la madre.
Discusión: En este caso nos interesa destacar la rareza del cuadro, su presentación como EQV, la facilidad del diagnóstico mediante una simple extensión de sangre periférica, el diagnóstico diferencial con otras enfermedades de depósito que cursan con ictiosis y el tratamiento, que es dietético, con restricción de grasas y uso de ácidos grasos de cadena media. La evolución con este régimen en nuestro paciente está siendo muy satisfactoria. Chanarin-Dorfman. Ictiosis. Depósito de lípidos neutros. Eritroqueratodermia variabilis. Gen NCIE2.
Displasia ectodérmica hipohidrótica ligada al cromosoma X: descripción de un caso femenino esporádico en mosaico
A. Toll, F. Gallardo y R.M. Pujol
Servicio de Dermatología. Hospital del Mar. Barcelona. España.
Introducción: La displasia ectodérmica hipohidrótica (DEH) se caracteriza por hipotricosis, hipodoncia, onicodisplasia e hipohidrosis. La incapacidad para transpirar puede provocar episodios de hipertermia con riesgo cerebral y/o vital. La forma ligada al cromosoma X es la forma más frecuente de DEH. Alrededor de dos tercios de las mujeres portadoras pueden manifestar algunos signos clínicos de DEH. Presentamos el caso esporádico de una mujer con una forma en mosaicismo de DEH ligada al cromosoma X. La prueba del yodo-almidón se mostró útil para evidenciar la enfermedad.
Caso clínico: Una mujer de 22 años nos consultó por presentar alopecia y un trastorno de la sudoración desde la infancia. No existían antecedentes familiares de trastornos similares. La paciente había sido diagnosticada de displasia ectodérmica durante la infancia debido a alteraciones en la dentición (hipodoncia y dientes cónicos). La exploración evidenció un pelo claro y ralo en cuero cabelludo y cejas. Las uñas eran normales. El signo más relevante observado fue la presencia de áreas lineales de piel pálida normal con presencia de anejos entremezclada con áreas de piel deprimida, seca en extremidades y piernas. Se realizó el test de yodo-almidón en la espalda de la paciente, que puso de manifiesto una distribución de las glándulas sudoríparas siguiendo las líneas de Blaschko. La biopsia cutánea mostró una pérdida segmentaria de glándulas ecrinas.
Comentarios: Los varones afectados de DEH ligada al cromosoma X muestran todas las manifestaciones del síndrome mientras las mujeres desarrollan una expresión fenotípica variable. En las mujeres heterozigotas la inactivación al azar de uno de los dos cromosomas X durante la embriogénesis se manifiesta con la presencia de áreas normales y anormales siguiendo las lineas de Blaschko. El caso que presentamos puede ser debido a una mutación poszigótica espontánea. A pesar de que la distribución siguiendo las líneas de Blaschko de las portadoras puede observarse en algunos casos a simple vista, la sencilla técnica de yodo-almidón puede ayudar a evidenciar estos casos, permitiendo iniciar los estudios de biología molecular adecuados.
Osteogénesis imperfecta tipo II: asociación con defectos del sistema nervioso central y/o defectos cardiovasculares
N. Baena1, E. Gabau2, J.C. Ferreres3, R. Bella3, F. Mellado4, M. Corona4, R.M. Perich2, C. Martín5 y M. Guitart1
Unidad de Diagnóstico Prenatal. 1Servicio de Laboratorio. 2Servicio de Pediatría. 3Servicio de Patología. 4Servicio de Ginecología y Obstetricia. 5Centro de Diagnóstico por la Imagen. Corporación Parc Taulí. Sabadell. Barcelona. España.
La osteogénesis imperfecta (OI) tipo II es un grupo heterogéneo de alteraciones causadas por una mutación en uno de los genes del colágeno tipo I (COL1A1 y COL1A2). La prevalencia es de 1/50.000 recién nacidos. El patrón de herencia es autosómico dominante, en algunas familias se sospecha de un mosaicismo en la línea germinal, y también se han descrito patrón de herencia autosómica recesiva. Se presentan 2 casos de OI tipo II en la misma familia asociados a defectos del SNC y/o cardiopatías diagnosticados prenatalmente mediante ecografía y RM en el segundo trimestre de la gestación. Pareja consanguínea con hija anterior sana. En el segundo embarazo se detecta a las 20 semanas de gestación una osteodisplasia asociada a defectos de SNC y arteria umbilical única. Tras el consejo genético la pareja decide interrumpir la gestación. El análisis anatomopatológico confirma que se trata de una OI tipo II asociada a una agenesia del cuerpo calloso, malformación de la fosa posterior y una arteria umbilical única. En el siguiente embarazo a las 14 semanas de gestación se observan defectos esqueléticos y una arteria umbilical única. Tras la interrupción voluntaria del embarazo se diagnostica una OI tipo II asociada a agenesia del cuerpo calloso, malformación cerebelosa, y defectos cardíacos: CIV submembranosa, atresia de válvula pulmonar e hipoplasia del tronco pulmonar, arteria umbilical única, y micropene e hipospadias escrotal. La malformaciones del SNC y cardiovasculares se hallan raramente asociadas a la OI tipo II. Se revisan los mecanismos patogénicos citados en la literatura para explicar dicha asociación.
Agradecimientos: Este trabajo ha sido financiado parcialmente por la Red de Enfermedades Raras (REpIER).
Síndrome cromosoma 20 en anillo e Hipomelanosis de Ito
R. Domingo-Jiménez1, I. López2, H. Alarcón1, C. Casas-Fernández1 y A. Puche-Mira1
1Sección de Neuropediatría. 2Sección de Citogenética. Instituto de Bioquímica Clínica y Genética. Hospital Universitario Virgen de la Arrixaca. Murcia. España.
Introducción: El síndrome de cromosoma 20 en anillo es una anomalía cromosómica infrecuente, caracterizada por epilepsia refractaria, retraso psicomotor, alteraciones de comportamiento y rasgos dismórficos. No se han comunicado alteraciones dermatológicas. Presentamos el caso de un niño con epilepsia, cromosoma 20 en anillo en mosaico y manchas hipopigmentadas características de la hipomelanosis de Ito.
Caso clínico: Niño de 7 años, sin antecedentes personales o familiares de interés. Enfermedad actual: crisis epilépticas, convulsivas y no convulsivas. Exploración: manchas hipocrómicas en las extremidades inferiores, siguiendo distribución de líneas de Blaschko, resto de exploración general y neurológica normal. Exámenes complementarios: EEG: actividad paroxísticas de predominio frontal derecho; RM cerebral normal. Cariotipo: cromosoma 20 en anillo en el 10 % de las metafases en sangre y en mayor proporción en fibroblastos de piel hipopigmentada. Evolución: epilepsia polimorfa de difícil control. Alteración conductual. Sin retraso mental.
Comentarios: El síndrome cromosoma 20 en anillo es una entidad bien definida clínica y neurofisiológicamente, constituye un tipo identificable de epilepsia. La pérdida de regiones teloméricas, donde se sitúan genes relacionados con canalopatías epilépticas, se cree que es el origen de la epilepsia. Las zonas de hipopigmentación que presenta este paciente son compatibles con hipomelanosis de Ito; cada vez hay más evidencias de que esta entidad no es una enfermedad, sino la manifestación de diferentes estados de mosaicismo. Se han publicado 2 casos de hipomelanosis de Ito y trisomía 20; sin embargo, no hemos encontrado casos asociados a cromosoma 20 en anillo.
Amioplasia congénita: una entidad con buen pronóstico
M. del Campo, R. Porta, V. Molina, C. Bonjoch y A. Muñoz
Institut Universitari Dexeus. Barcelona. España.
Introducción: El objetivo de este estudio es recordar, a propósito de un caso clínico, la amioplasia congénita como una forma de artrogriposis esporádica y con buen pronóstico.
Caso clínico: Primera gestación de madre joven y sana, que cursa sin complicaciones obstétricas. Las ecografías fetales ponen de manifiesto anomalías posicionales fijas de extremidades, que persisten en los controles hasta el final de la gestación: EID en extensión fija y extremidad inferior en flexión fija, sin movilidad de rodilla y tobillo, y pies equinovaros bilaterales; las últimas ecografías ponen de manifiesto también extensión fija del codo en ESI y muñeca doblada con desviación cubital. No existen anomalías asociadas de otros órganos. El parto tiene lugar a las 38 semanas por cesárea electiva, naciendo una niña con test de Apgar 9/10, peso de 2.980 g, y aspecto sano, y que presenta anomalías posicionales de articulaciones concordantes con las descritas prenatalmente. Además de las anomalías de extremidad inferior, la ESI muestra anomalías simulando una parálisis de plexo braquial. Presenta un curso clínico sin problemas, y se inicia fisioterapia desde los primeros días. En la actualidad tiene 5 meses de vida, es controlada por equipo multidisciplinar (traumatólogo, fisioterapeuta, neurólogo), y la evolución de sus anomalías musculoesqueléticas es satisfactoria. Muestra un neurodesarrollo normal.
Comentario: La amioplasia congénita es, en la práctica clínica, la forma más frecuente de artrogriposis (30 % de casos). Su incidencia es de alrededor de 1/10.000 recién nacidos. El neonato afectado muestra una apariencia saludable y un peso normal, suele mostrar malformaciones vasculares capilares mediofaciales y múltiples articulaciones fijas, simétricas, con posición y distribución atípicas. Es muy característica la posición de las extremidades superiores: hombros en aducción y rotados medialmente, codos extendidos o flexionados, antebrazos en pronación, muñecas dobladas con desviación cubital y dedos flexionados. Las extremidades inferiores, también a menudo comprometidas, muestran rodillas flexionadas o extendidas y pies equinovaros o calcaneovalgos. Aunque se han descrito casos de atresia intestinal y gastrosquisis, no hay alteraciones de otros sistemas. Su causa no se conoce, aunque se postula la necrosis de la médula espinal fetal por hipotensión sistémica. Es importante conocer esta entidad porque es una afectación esporádica con pocas probabilidades de recidivas y cursa con inteligencia normal. El pronóstico funcional con tratamiento adecuado (fisioterapia, ortopedia) es bueno. En este caso, la precisión de la ecografía prenatal, que limitó las anomalías a EE, facilitó la sospecha diagnóstica prenatal, la anticipación y la preparación psicológica de los padres, que se vio apoyada con la visita prenatal a genetista clínico y neonatólogo.
Síndrome de Neu-Laxova y síndrome de Cofs: ¿Candidatos paralumping? Presentación de un caso y revisión de la literatura médica
E. Bermejo, M. Calvo, M. García González, M. Alegre, I. Vila, L. Cuevas, F. López, J. Mendioroz, E. Rodríguez-Pinilla y M.L. Martínez-Frías
ECEMC, CIAC, Instituto de Salud Carlos III, Madrid (EB, LC, FL, JM, ERP, MLMF); Serv. Pediatría (MC, MGG) y Servicio de Obstetricia y Ginecología (MA, IV). Hospital Comarcal de Figueres. Girona. Departamento de Farmacología. Facultad de Medicina. Universidad Complutense. Madrid. España.
El síndrome de Neu-Laxova se caracteriza por la presencia de retraso del crecimiento intrauterino, microcefalia profunda, edema generalizado, contracturas de miembros, alteraciones del SNC y, con frecuencia, piel de aspecto ictiósico. Es un síndrome letal y tiene herencia autosómica recesiva. El síndrome de COFS, cuyo nombre responde al acrónimo de sus manifestaciones principales en inglés (Cerebral, Ocular, Facial y Skeletal) es también autosómico recesivo y entre los criterios diagnósticos mayores se incluye la microcefalia, alteraciones oculares (catarata y microftalmía, fundamentalmente), faciales (puente nasal elevado, labio superior prominente, boca y barbilla pequeñas, micrognatia), cifosis/escoliosis, contracturas de miembros, luxación/displasia de cadera, pies en mecedora y otras alteraciones esqueléticas. Presentamos el caso de una niña nacida a las 35 semanas de gestación, fruto de la segunda gestación (el embarazo anterior finalizó en un aborto espontáneo) de padres sanos, no consanguíneos. La madre tenía 32 años cuando nació la niña y el padre 35. En cuanto a la historia obstétrica, la madre tuvo una infección de orina sin fiebre en el primer trimestre, tratada con ciprofloxacino e hiperemesis gravídica en los primeros 3 meses, no tratada. La madre refiere que no percibió movimientos fetales en ningún momento y se observó polihidramnios. No hubo exposiciones conocidas a teratógenos. El cariotipo realizado tras practicar funiculocentesis fue aparentemente normal (46,XX). La somatometría al nacimiento fue: 1.160 g, 37,5 cm y perímetro cefálico (PC) de 26 cm, medidas todas que se sitúan por debajo del P3 para su edad gestacional. En la exploración física se observó edema en cuero cabelludo y extremidades, hipercifosis dorsal, orejas de implantación baja, hendiduras palpebrales cortas, microrretrognatia, fisura palatina, cuello corto, flexión de hombros, codos, muñecas, dedos, caderas (con limitación a la abducción) y rodillas, pies talos, xifoides prominente e hipoventilación bilateral en la auscultación por debilidad de los movimientos respiratorios. Precisó administración de flujo continuo de oxígeno y a los 30 min se observó un descenso progresivo de la saturación de hemoglobina, falleciendo la niña a la hora de vida. Los datos adicionales obtenidos de la autopsia son los siguientes: catarata bilateral, signos de hipoxia cerebral con pérdida neuronal y calcificaciones del tálamo y focos de ictiosis cutánea. En la presentación se discutirá el diagnóstico diferencial entre el síndrome de Neu-Laxova y el fenotipo COFS, así como la posibilidad de que ambos sean considerados como una única entidad nosológica.
