
Sr. Editor:
Desde finales de la década de 1960 se ha observado una fuerte asociación entre el déficit de antitrombina III (AT III) y la trombosis. Esta deficiencia puede ser de tipo cuantitativo o cualitativo, y se clasifica de acuerdo con los rasgos fenotípicos.
El gen de la AT III se encuentra en la región 23–25 del brazo largo del cromosoma 1, y sigue un patrón de herencia autosómico dominante. En los estudios de pacientes con déficit de AT III se han encontrado mutaciones en el sitio reactivo y en el lugar de unión a la heparina1; la prevalencia de trombosis es variable. Se reconoce un tipo I de déficit de antitrombina, caracterizado por una disminución de AT III medida con técnicas funcionales e inmunológicas y un tipo II de déficit de AT III caracterizado por un déficit funcional. El tipo II se divide en tres subtipos. El primero, conocido como RS, corresponde a una variante de la molécula de AT, la cual tiene un defecto en el centro activo. El segundo se denomina HBS, un defecto que afecta al lugar de unión de la heparina y, por último, existe la deficiencia funcional múltiple conocida como PE (pleiotropic effect)2,3.
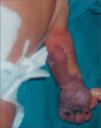
Se presenta el caso de un recién nacido que presentaba zona eritematosa en la cara anterior del antebrazo izquierdo.
Es fruto de un primer embarazo de 41 semanas de duración. La madre tuvo hemorragia en el primer mes de gestación. El parto fue de inicio espontáneo, posteriormente inducido por sufrimiento fetal agudo. Apgar 3-7-9, reanimación tipo III. Los antecedentes maternos se desconocían en el momento del parto.
Ingresó en la unidad neonatal, procedente de paritorio, por distrés respiratorio leve. En la exploración inicial presentaba una zona roja sugerente de angioma, en la cara anterior del antebrazo, de 3 × 1cm. Después de 14h de vida, la lesión ocupaba toda la cara anterior del antebrazo y mano izquierdos y su aspecto sugería la existencia de patología vascular. En la exploración se apreciaba ausencia de pulso axilar, humeral y distal de dicha extremidad. El estudio con eco-Doppler confirmó trombosis axilo-subclavio-humeral izquierda de todo el eje, que había comenzado probablemente, en la zona subclavio-humeral. Los hallazgos con eco-Doppler explicaron que, al no tener colateralidad de suplencia suficiente, fuera creciendo una zona de prenecrosis desde los dedos hasta mitad del antebrazo izquierdo en las 24–48h siguientes (fig. 1).
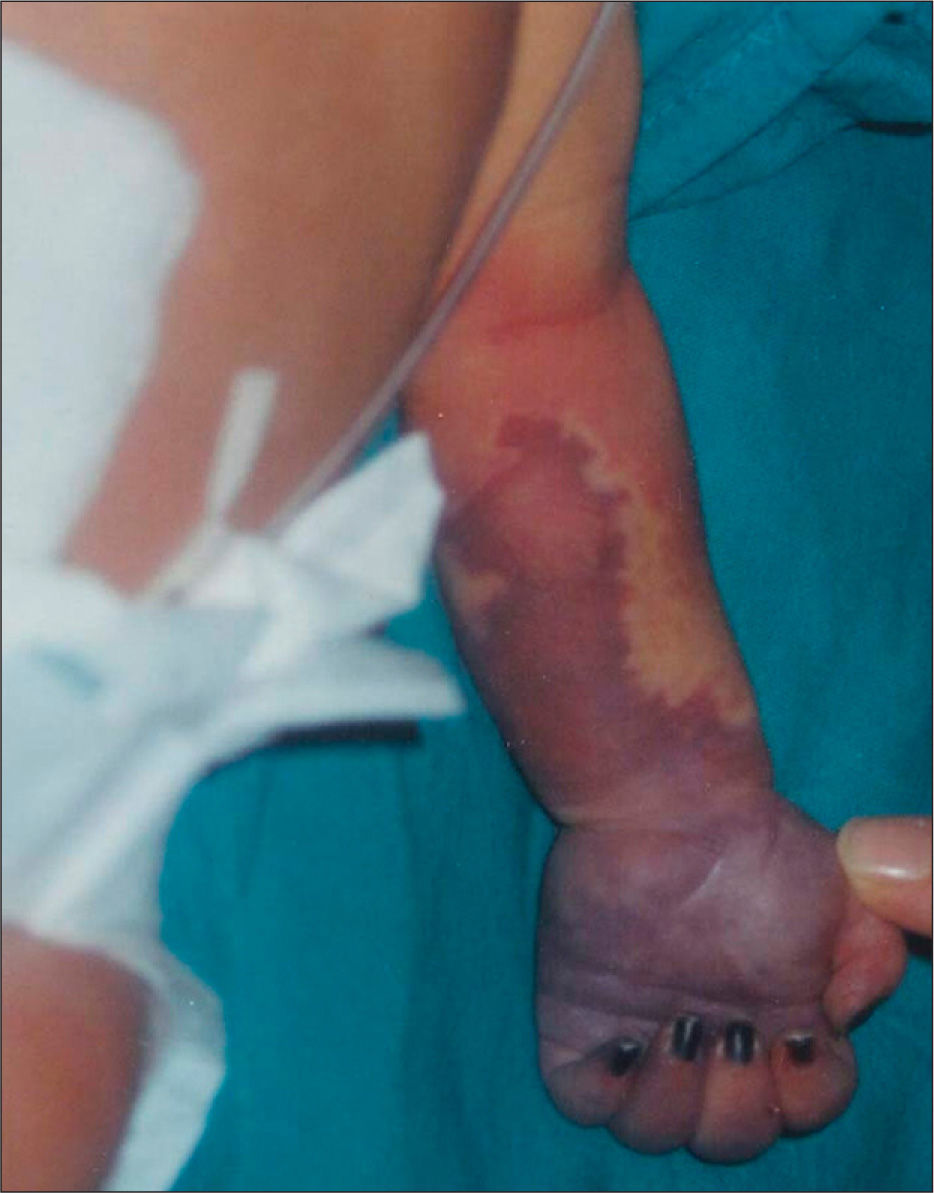
Ante la evidencia de patología trombótica se realizaron pruebas de coagulación, y presentaba actividad de protrombina: 25%, índice normalizado internacional (INR): 3,26,7; tiempo de tromboplastina parcial activado (TTPA): 56,6s; AT III: 36%. Se confirmaba el déficit de AT. Se inició tratamiento con concentrado de AT III y heparina sódica en dosis anticoagulantes, ajustando las dosis diarias previo estudio de coagulación y cifras de AT III. Se mantuvieron unas cifras de AT del 70 %, y mejoró la perfusión de forma progresiva del brazo y tercio superior de antebrazo izquierdos, pero la mitad inferior del antebrazo y la mano terminó necrosándose. Cuando se consiguió una buena delimitación de la zona necrótica se procedió a su amputación.
El estudio genético molecular del gen protrombina, para detectar la presencia de una mutación concreta en la posición 20210, y el estudio del factor V para detectar mutación en la posición 1691 y reacción en cadena de la polimerasa (PCR) en hibridación con sondas específicas, fueron negativos. Sí presentaba homocigosis para la mutación del gen de la metilentetrahidrofolato reductasa en la posición 677.
El estudio amplio de los parámetros de la antitrombina circulante en plasma citratado presentó una actividad anticoagulante de antitrombina plasmática normal. Las concentraciones antigénicas mediante electroforesis en geles de agarosa (método de Laurell) fueron normales y estudios conformacionales mediante Western blot no evidenciaron formas conformacionales diferentes, ni mayor presencia circulante de formas latente o polimérica de antitrombina.
En el análisis molecular se amplificaron por PCR los 6 exones y regiones flanqueantes del gen de la antitrombina, observando una mutación concreta missense en heterocigosis localizada en el exón 6 (13296G-A), responsable del cambio R393H que afecta al residuo P1 del centro reactivo de la molécula. Estudios de restricción en el paciente confirmaron la presencia de la mutación. Mediante PCR-ASRA se confirmó al paciente como afectado de esta alteración genética. Esta mutación ha sido previamente caracterizada, conocida con el nombre de AT Glasgow.
En el estudio familiar se encontró déficit de AT III y factor V de Leyden en la familia materna: la bisabuela materna falleció por embolismo cerebral; el abuelo materno con déficit de AT III; la abuela materna con déficit de factor V de Leyden y consanguinidad entre ellos. La madre tenía ambos déficit diagnosticados tras la gestación, y fue también homocigota para la mutación del gen de la metilentetrahidrofolato reductasa en la posición 677 y sin antecedentes trombóticos.
La estimación de la prevalencia de la trombosis en pacientes con déficit de AT III demuestra que no todos sufren trombosis, por lo que habría que investigar otros probables factores asociados4,5.
En cuanto al riesgo de mortalidad, aunque se asocia a un alto riesgo de trombosis venosa profunda, no parece aumentarla. Por ello, no está justificada la profilaxis anticoagulante en todos los tipos de deficiencia, debiendo valorarse únicamente ante las manifestaciones trombóticas, o en determinadas situaciones de mayor riesgo, como el embarazo, cirugía e inmovilización.
In memoriamAl Dr. Samaniego. En recuerdo de un gran profesional y mejor compañero.
