


Sr. Editor:
Las cromosomopatías son una de las principales causas de malformaciones mayores en el recién nacido y, además, en la actualidad suponen un causa relevante de muerte en niños menores de un año 1,2. La duplicación parcial del brazo corto del cromosoma 5 es una anomalía poco frecuente 3. La expresión clínica es muy variada aunque ya conocida. Se describe el caso clínico de un recién nacido con rasgos faciales dismórficos y cardiopatía congénita.
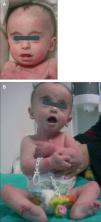
Recién nacida fruto de un embarazo controlado, de padres sanos, no consanguíneos y sin historia familiar de malformación fetal. Parto a término (38 + 3 semanas) mediante cesárea urgente por pérdida de bienestar fetal. El test de Apgar al minuto de vida fue de 8 y, a los 5 y 10 min de 9. Los datos somatométricos al nacer fueron: peso 2.085 g (percentil < 10), talla 43,4 cm (percentil < 10) y perímetro de cráneo 33,5 cm (percentil 20). En la exploración física destacaba: macrocefalia, un llanto agudo y los siguientes rasgos faciales: microrretrognatia, hendiduras palpebrales oblicuas ascendentes, hipertelorismo, lóbulos auriculares pequeños, orejas de implantación baja y antevertidas y nariz en silla de montar. Además, se observaban dedos largos y pies varos reducibles (fig. 1A y B). Desde el punto de vista neurológico, mostraba un débil reflejo de succión y prensión, así como hipotonía generalizada. A la auscultación cardíaca presentaba un soplo sistólico multifocal 2/6.
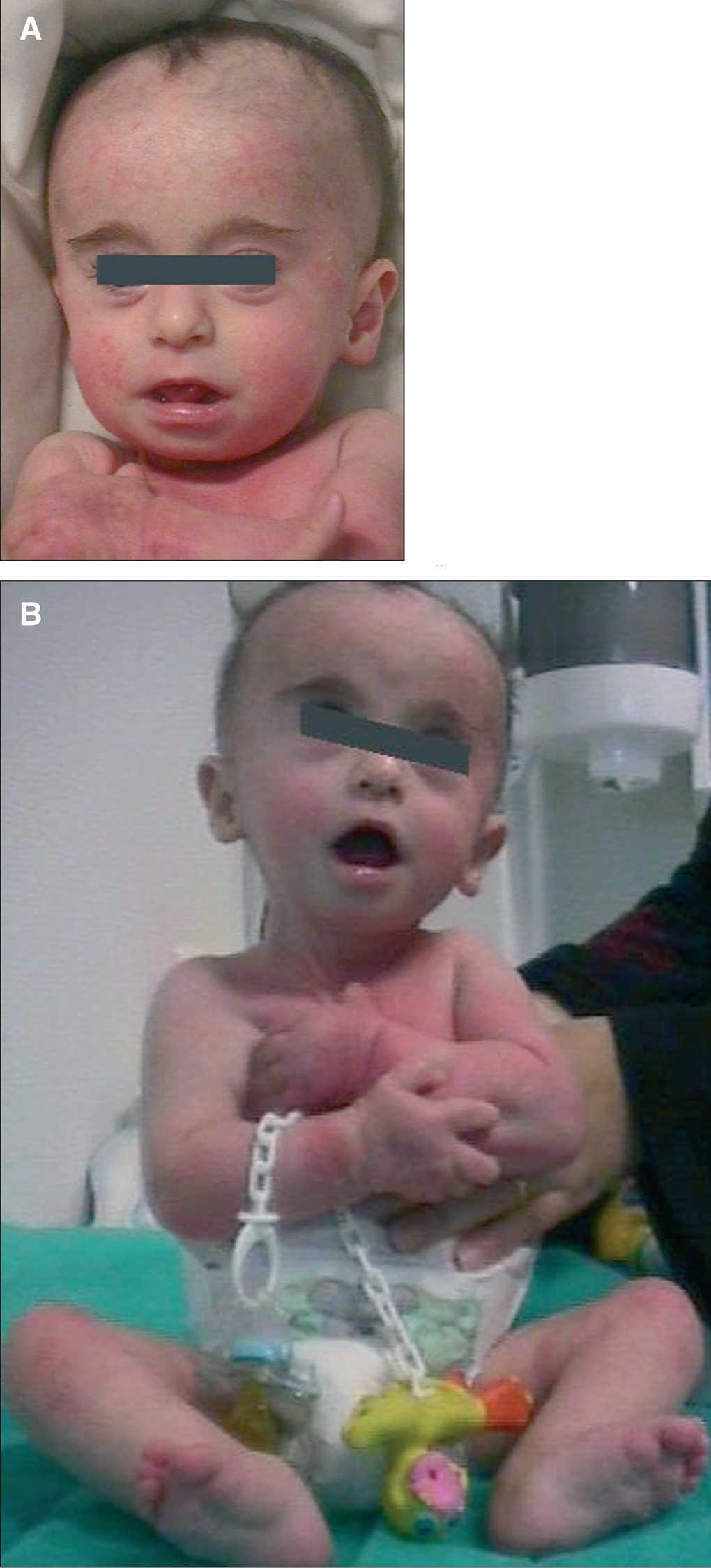
Figura 1A y B. Rasgos morfológicos de la paciente (trisomía parcial 5p).
Entre las pruebas diagnósticas solicitadas la ecocardiografía realizada a las 12 h de vida demostró una comunicación interventricular subaórtica mediana, comunicación interauricular mediana tipo ostium secundum y ductus mediano. El tránsito esofagogástrico evidenció incoordinación velopalatina y ligera compresión del tercio distal del esófago, relacionado con el aumento de la aurícula izquierda. El estudio genético realizado sobre los linfocitos de sangre periférica mediante cultivo, choque hipotónico y evaluación microfotográfica estableció el diagnóstico definitivo de trisomía parcial del cromosoma 5. Se obtuvo la siguiente fórmula: 46,XX, t(5;13). Se realizaron técnicas de hibridación in situ aplicando varias sondas en las se observó fluorescencia completa de los dos cromosomas normales y de la región duplicada del brazo corto: 46,XX, t(5;13)ish(wcp15x3, D5Fx3, BSP5p15.2-13x3). La técnica de hibridación in situ demostró que todas las bandas del brazo corto del cromosoma 5 estaban afectadas. Se estudiaron los cariotipos de los padres, que fueron normales. A la semana de vida comenzó a presentar síntomas de taquipnea, tiraje subcostal e intercostal y taquicardia. La ecocardiografía realizada en ese instante reveló datos de sobrecarga de cavidades derechas con dilatación e hiperaflujo pulmonar y se instauró tratamiento con furosemida. La ecocardiografía realizada al mes de vida evidenció una orejuela de la aurícula izquierda muy dilatada y una coartación aórtica leve. Ante estos hallazgos se inició tratamiento con digoxina que mantiene en la actualidad. En los 8 meses siguientes se observó un lento progreso ponderal; recibe alimentación enteral por sonda nasogástrica. Destaca un moderado retraso psicomotor, eccema atópico severo e infecciones respiratorias virales recurrentes. La resonancia magnética realizada a los 4 meses de vida no demostró signos definitivos de alteración de la migración neuronal.
La trisomía parcial del brazo corto del cromosoma 5 es una alteración cromosómica poco frecuente. Dicha cromosomopatía puede ser debida a una amplia variedad de mutaciones cromosómicas lo cual deriva en un amplio abanico de síntomas y signos 4,5. Si bien las características morfológicas observadas en este caso coinciden en su mayoría con lo descrito anteriormente en la bibliografía 3,6-8, nuestra paciente supone el primer caso encontrado en la literatura médica de trisomía parcial del cromosoma 5p debida a la translocación 5;13 de novo. El diagnóstico definitivo se realiza mediante el estudio genético realizado sobre linfocitos de sangre periférica con cultivo, choque hipotónico y evaluación microfotográfica. La región triplicada puede ser desde una subbanda G hasta la totalidad del cromosoma 5 4. En el caso presentado se obtuvo la fórmula 46,XX, t(5;13). La presencia del defecto 5p13-15 se ha relacionado con retraso mental 9 descrito también en nuestra paciente. Pese a que lo más frecuente en la literatura médica son los casos de trisomía 5p secundarios a la herencia de translocaciones balanceadas 3, en el caso expuesto el cariotipo de los padres era normal. El pronóstico depende en mayor medida de las alteraciones neurológicas: importante retraso psicomotor y convulsiones refractarias 10. No obstante, se desconoce la supervivencia de la trisomía parcial 5p a largo plazo. Nuestra paciente al año de vida no ha presentado sintomatología comicial. En la actualidad no existe una terapia génica aplicada a este defecto cromosómico, por lo que el tratamiento en todos los casos será sintomático 7. Se realizará consejo genético ante la posibilidad de posibles embarazos.
Correspondencia: Dra. S. Marcos Alonso.
Servicio de Neonatología. Departamento de Pediatría.
Hospital Clínico Universitario de Santiago de Compostela.
La Choupana, s/n. 15706. Santiago de Compostela. España.
Correo electrónico: soniamarcosalonso@hotmail.com
