



Sr. Editor
La fibrodisplasia osificante progresiva (FOP), miositis osificante o enfermedad de Munchmeyer es una alteración genética infrecuente del tejido conjuntivo. La mayoría de los pacientes corresponden a mutaciones de novo1, aunque existen casos de transmisión dominante. Su incidencia es de 1/2.000.000 individuos y no muestra predilección sexual, racial o étnica. Se caracteriza por: a) anomalías esqueléticas, fundamentalmente hallux valgus congénito; b) osificación heterotópica de la musculatura estriada, fascias, tendones y ligamentos, y c) progresión del cuadro clínico 2. La inmovilización de grandes articulaciones le confiere mal pronóstico funcional y la de la caja torácica origina a menudo un fallo respiratorio restrictivo.
Se describen a continuación 2 pacientes con el propósito de ayudar al reconocimiento precoz de esta enfermedad invalidante.
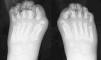
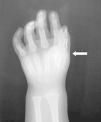
Caso 1. Varón sin antecedentes de interés que en julio de 2003, a los 30 meses de edad, ingresó por una tumoración pequeña, mal definida y dolorosa en región cervical derecha, que coincidiendo con la varicela se extendió hacia la escápula. Estaba afebril y tanto la analítica como el cariotipo fueron normales. Se evidenció hallux valgus (fig. 1), clinodactilia y una calcificación de 2 x 1 cm en la eminencia tenar de la mano derecha (fig. 2). La ecografía y la resonancia magnética revelaron engrosamiento de la fascia. La biopsia resultó compatible con fascitis nodular versus seudotumor inflamatorio. Por la sospecha de FOP, en agosto inició tratamiento con alendronato 20 mg/semana de forma continuada y con corticoides e ibuprofeno en los empeoramientos clínicos. En junio de 2004 la enfermedad había progresado y se evidenciaban masas de consistencia dura a lo largo de la espalda, en regiones paravertebrales y bajo la axila derecha, con importante limitación para la movilidad del brazo, que apenas podía separar del tronco y de la columna cervical, que sólo conservaba el movimiento de flexión.
Figura 1. Hallux valgus.
Figura 2. Clinodactilia y calcificación en eminencia tenar.
Caso 2. Niña diagnosticada de FOP a los 3 años por limitación para la flexión de rodilla derecha y calcificaciones en tejidos blandos, hallux valgus y clinodactilia. Los antecedentes familiares carecían de interés. Nos fue remitida desde el servicio de Ortopedia en 1994, a los 13 años, por dolor mecánico en ingle derecha. La exploración mostró contractura en extensión y ausencia de movilidad del codo y la rodilla derechos, flexo de 40° no reducible en la cadera derecha, e imposibilidad para las rotaciones, aunque flexionaba hasta 90º en la cadera izquierda. La capacidad para deambular estaba muy limitada. La radiografía de caderas demostró calcificaciones de tejidos blandos (fig. 3). Desde los 15 años se objetivó trastorno ventilatorio restrictivo moderado. El cuadro progresó a pesar de tratamiento con alendronato 10 mg diarios y de la administración de indometacina y corticoides en los episodios de dolor y tumefacción. A los 20 años fue perdida para el seguimiento.

Figura 3. Calcificaciones en partes blandas.
El diagnóstico de la FOP es esencialmente clínico y radiológico 3. Afecta a niños menores de 10 años, con alteraciones esqueléticas (hallux valgus congénito, acortamiento del primer metacarpiano, clinodactilia) que con o sin antecedente traumático, presentan masas dolorosas de características seudoinflamatorias, las cuales se trasforman en tejido óseo maduro mediante osificación endocondral. La osificación heterotópica progresa con un patrón secuencial en dirección cefalocaudal, proximodistal y axialapendicular 4. La mejor técnica de imagen para establecer el diagnóstico es la radiografía convencional. La tomografía computarizada, los ultrasonidos y la resonancia magnética no son imprescindibles 5. La histología es poco relevante y se deben evitar las biopsias, ya que representan un estímulo para nuevas osificaciones 6, aunque a veces, como en nuestro primer caso, se realizan para descartar infecciones y neoplasias.
En pacientes con FOP se ha descrito sordera, alopecia y más rara vez rasgos dismórficos faciales 7.
La fisiopatología de la enfermedad no está claramente establecida. Se ha demostrado la sobreexpresión de una proteína formadora de hueso, bone morphogenetic protein-4 (BMP4) 8,9, y se desconoce el mecanismo molecular por el que se produce 10. Además, existe una escasa producción de los antagonistas de BMP4. La mutación del gen que codifica la síntesis de BMP4 se ha excluido como responsable del proceso.
Hasta el momento ningún fármaco ha logrado alterar la historia natural de la enfermedad, cuya evolución es muy variable entre diferentes individuos e incluso en un mismo paciente 11-13. Pese a que no existen evidencias respecto a la prevención y el tratamiento idóneos, parece razonable evitar traumatismos, como inyecciones intramusculares y usar antiinflamatorios y corticoides durante cortos períodos en los episodios de dolor e inflamación. Pamidronato, colchicina, talidomida entre otros medicamentos han sido utilizados en casos aislados, con resultados poco concluyentes. Nuestros pacientes recibieron alendronato, sin conseguir detener la progresión del cuadro. Los agentes de ingeniería genética anti-BMP4, no disponibles aún para uso clínico, parecen abrir expectativas esperanzadoras 14.
Por último, la Asociación Internacional de Fibrodisplasia Osificante Progresiva (http://www.ifopa.org) ofrece una revisión actualizada, incluyendo las líneas de investigación que hoy están abiertas 15.
Correspondencia: Dra. B. Pérez-Seoane Cuenca.
Unidad de Reumatología Pediátrica.
Hospital Universitario La Paz.
P.º de la Castellana, 261. 28046 Madrid. España.
Correo electrónico: reumaped.hulp@salud.madrid.org
