
Las ceroidolipofuscinosis neuronales (CLN) representan un grupo de enfermedades lisosomales hereditarias de herencia autosómica recesiva, de presentación más frecuente durante la niñez, caracterizadas neuropatológicamente por acumulación de lipopigmentos autofluorescentes en los lisosomas de neuronas y otras células. Clínicamente se presentan con pérdida de las habilidades psicomotoras adquiridas, incoordinación motora, ataxia, pérdida de la visión, cambios de conducta, convulsiones de difícil tratamiento asociadas a mioclonías y una corta expectativa de vida. En la actualidad, se conocen 10 formas genéticamente distintas de esta enfermedad, entre ellas la forma infantil tardía donde las manifestaciones clínicas aparecen entre el segundo y cuarto año de vida. El gen responsable de la enfermedad es el TPP1 ubicado en 11p15 y codifica la enzima tripeptidil peptidasa 1.
Pacientes y métodosSe estandarizó la técnica para el diagnóstico enzimático de la ceroidolipofuscinosis neuronal infantil tardía a través de sangre seca en papel de filtro en 76 individuos sanos en edad preescolar y adulta de población venezolana. La actividad enzimática de la TPP1 fue determinada en 9 pacientes con diagnóstico clínico de ceroidolipofuscinosis infantil tardía 2 (CLN2).
ResultadosSeis pacientes mostraron valores de actividad muy por debajo del rango establecido (0,11-0,45 nmol/mancha) para los controles sanos en edad preescolar, confirmando el diagnostico enzimático. Tres de los 14 padres estudiados presentaron valores en el rango de heterocigotos.
ConclusionesEl diagnóstico enzimático de CLN2 a través de la determinación de la actividad enzimática de la enzima TPP1 mediante la técnica de sangre seca en papel de filtro permite un diagnóstico rápido, sencillo, económico y confiable.
Neuronal ceroid lipofuscinoses are a group of inherited autosomal recessive lysosomal diseases, most commonly found in infancy. These are neuropathologically characterised by accumulation of an autofluorescent lipopigment in neurons and other cells. This condition is clinically characterised by loss of motor and cognitive skills, lack of motor coordination, ataxia, progressive visual impairment, behavioural changes; seizures of difficult to manage seizures, particularly myoclonic, and premature death. Ten clinical forms have been described, one of which is late infantile where clinical signs begin between two and four years. The gene responsible for this disease is located at 11p15 locus, and the enzyme encoded by this gene is the tripeptidyl peptidase 1.
Patients and methodsWe standardised the technique for the enzymatic diagnosis of late infantile neuronal ceroid lipofuscinoses from dried blood on filter paper card in 76 healthy individuals adults and children in order to establish a normal range in the Venezuelan population. The tripeptidyl peptidase activity was also determined in 9 patients with a clinical diagnosis of late infantile neuronal ceroid lipofuscinoses.
ResultsSix of the samples showed activity lower than the lowest control value (0.11 to 0.45 nmol/spot) from healthy controls of infantile age, confirming the enzymatic diagnosis. Three of the 14 parent samples analysed showed values in the heterozygote ranges.
ConclusionsThe enzymatic diagnosis of late infantile neuronal ceroid lipofuscinoses from dried blood on filter paper card is a rapid, easier, less expensive and accurate molecular diagnosis tool.
Las ceroidolipofuscinosis neuronales (CLN) representan un grupo de enfermedades lisosomales hereditarias neurodegenerativas caracterizadas por marcado y progresivo deterioro neurológico, con pérdida de las habilidades psicomotoras adquiridas, incoordinación motora, ataxia, pérdida de la visión, convulsiones, mioclonías y muerte1. Las formas infantiles de las CLN representan la enfermedad neurodegenerativa más común en niños y son heredadas de modo autosómico recesivo1-4. La incidencia mundial estimada para todos los tipos es de 1:100.000 nacidos vivos5,6. Originariamente, las CLN fueron clasificadas, basándose en la edad de inicio de los síntomas clínicos, en 4 formas: infantil (CLN1), infantil tardía (CLN2), juvenil (CLN3) y adulta (CLN4)7. Actualmente, se conocen 10 formas diferentes de CLN de acuerdo al defecto del gen, las cuales son reconocidas como enfermedades de depósito lisosomal. El estudio patológico reporta la presencia de lipopigmentos ceroide y lipofuscina8. Desde el punto de vista ultraestructural, estos lipopigmentos se presentan como depósitos granulares osmiofílicos (GROD), cuerpos curvilíneos o como estructuras en huella dactilar, encontrados en los linfocitos circulantes y en las biopsias de piel o músculo9,10.
En la ceroidolipofuscinosis infantil tardía (CLN2) o enfermedad de Jansky-Bielschowsky las manifestaciones clínicas aparecen entre el segundo y cuarto año de vida. El gen responsable de este trastorno esta ubicado en el locus 11p15 y la proteína codificada por este gen es la enzima tripeptidil peptidasa 1 (TPP1), que escinde los tripéptidos del extremo N-terminal de las proteínas pequeñas antes de su degradación por otras proteasas lisosomales y el sustrato acumulado se presenta generalmente bajo la forma de cuerpos curvilíneos (CV)11.
Los estudios complementarias utilizados para el diagnóstico de las CLN (potenciales evocados visuales y auditivos, electroencefalograma, electrorretinograma y neuroimágenes) y estudios de anatomía patológica (microscopía electrónica), permiten confirmar el diagnóstico pero no distinguen entre sus diferentes formas clínicas. La edad de inicio, la evolución clínica, las neuroimágenes y los hallazgos anatomopatológicos no guardan una correlación específica para cada tipo y variante. Es por ello que los esfuerzos actuales están dirigidos inicialmente a la determinación de la actividad enzimática residual. La deficiencia de la actividad enzimática específica puede demostrar un estado homocigoto o heterocigoto compuesto para el gen mutante. Confirmada la deficiencia enzimática, el siguiente paso es el análisis molecular para la identificación de la mutación.
El objetivo de la investigación fue: establecer el rango de referencia de la actividad de la enzima TPP1 expresada en sangre seca en una población venezolana y demostrar la deficiencia de la actividad enzimática de la TPP1 en pacientes con diagnóstico clínico de CLN2.
Pacientes y métodosSe estudió a 9 pacientes con diagnóstico clínico de CLN2 infantil tardía (CLN2), de los cuales 3 fueron mujeres y 6 varones, entre 2 y 9 años de edad. A cada paciente se le elaboró su historia clínica genética y estudios complementarios. Se determinaron las actividades enzimáticas a cada uno de los pacientes y a sus padres. El grupo control estuvo conformado por 40 preescolar y 36 adultos voluntarios, sanos, de ambos sexos, sin antecedentes familiares de CLN ni cualquier otra enfermedad neurodegenerativa, malformaciones congénitas y/o enfermedades hereditarias.
El sustrato Ala-Ala-Phe-7-amido-4-methylcoumarin y los reactivos dimetil sulfóxido, triton X-100, acetato de sodio, cloruro de sodio, hidróxido de sodio, ácido tricloroacético, ácido acético y 4-metilcumarina fueron suministrados por Sigma-Aldrich.
La actividad enzimática fue determinada en gotas de sangre seca sobre papel de filtro calibrado (GSS) según la técnica descrita por Zoltan L et al.12 (2003). Las gotas de sangre seca fueron ponchadas por duplicado en placas de cultivo y eluídas en 20μl de buffer sustrato 0,25mM y 40μl de NaCl 9g/l. Posteriormente, se incubó a temperatura ambiente por 45min en un agitador de placa a 300rpm. Seguidamente, se incubó a 37°C por 45 h, en cámaras húmedas. La reacción se detuvo por adición de 440μl de buffer de parada pH 4,3 (ácido tricloroacético 0,1mol/l, NaOH 0,13mol/l, CH3COOH 0,1mol/l) en cada pozo. La fluorescencia liberada por cada una de las muestras fue medida a 355nm de excitación y 460nm de emisión en un lector Synergy HT reader (Bio-Tek, Vermont, EE. UU.).
Para descartar la fluorescencia proveniente de la hemoglobina se preparó en otros pozos muestras de sangre seca sin sustrato y se eluyó la hemoglobina durante 15min.
Los resultados obtenidos se corrigieron para los blancos para cada muestra y las cantidades de 4-metilcumarina, se calcularon a partir de la curva de calibración. La actividad enzimática se expreso en las unidades pertinentes (nmol/mancha).
Análisis estadísticoPara el análisis estadístico de los datos se utilizó el programa estadístico SPSS (Statistical Package for the Social Sciences) en su versión 15.0 para Windows (2006). Los datos fueron presentados mediante el uso de tablas de contingencia, con valores expresados en porcentajes o promedio±desviación estándar (DS). Se consideró el 95% como índice de confiabilidad estadística y como significativo un valor de p<0,05.
ConsentimientoLa investigación cumplió con todos los principios éticos concordantes con la Declaración de Helsinski (2004) para la investigación médica que involucre a seres humanos, material humano y datos de identificación. Igualmente, la investigación fue aprobada por el comité del Instituto de Genética Médica de la Facultad de Medicina de la Universidad del Zulia.
ResultadosSe analizó la actividad de TPP1 en 76 individuos sanos (40 niños edad preescolar y 36 adultos) y 9 pacientes de ambos sexos.
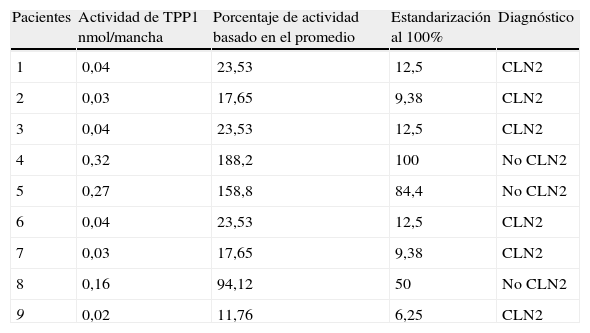
El rango de actividad enzimática de TPP1 para niños controles (n=40) fue de 0,11-0,45 nmol/mancha, con un promedio de 0,17±0,05 nmol/mancha. La tabla 1 muestra el porcentaje de actividad enzimática de la PPT1 en pacientes con CLN2 basado en el promedio de actividad enzimática de los controles. Con respecto a los pacientes con diagnóstico clínico de CLN2, se observó que 6 de 9 exhibieron valores de actividad de 0,02-0,04 nmol/mancha y promedio de 0,03 nmol/mancha, con valores estandarizados en base al 100% se encontraban entre el 6,25 y el 12,5%.
Actividad enzimática de tripetidil peptidasa 1 (TPP1) y porcentajes de actividad determinadas en gotas de sangre seca en papel en pacientes con diagnóstico clínico de CLN2
| Pacientes | Actividad de TPP1 nmol/mancha | Porcentaje de actividad basado en el promedio | Estandarización al 100% | Diagnóstico |
| 1 | 0,04 | 23,53 | 12,5 | CLN2 |
| 2 | 0,03 | 17,65 | 9,38 | CLN2 |
| 3 | 0,04 | 23,53 | 12,5 | CLN2 |
| 4 | 0,32 | 188,2 | 100 | No CLN2 |
| 5 | 0,27 | 158,8 | 84,4 | No CLN2 |
| 6 | 0,04 | 23,53 | 12,5 | CLN2 |
| 7 | 0,03 | 17,65 | 9,38 | CLN2 |
| 8 | 0,16 | 94,12 | 50 | No CLN2 |
| 9 | 0,02 | 11,76 | 6,25 | CLN2 |
Controles: n=40.
Promedio de actividad en niños controles: 0,17±0,05nmol/mancha.
Rangos: 0,11-0,45 nmol/mancha.
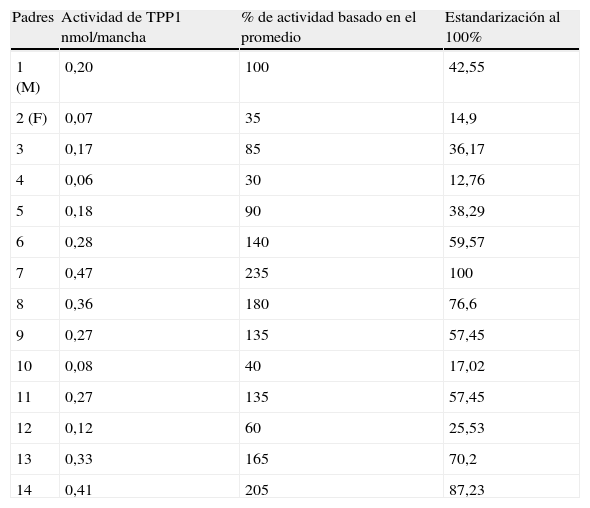
La actividad enzimática de TPP1 encontrada en padres de pacientes con CLN2 se muestran en la tabla 2; el rango de actividad enzimática en adultos sanos fue de 0,11-0,50 nmol/mancha con un promedio de 0,20 nmol/mancha±0,08. Los valores estandarizados en porcentajes se encontraban entre 12,76-100% de actividad enzimática.
Actividad enzimática de tripetidil peptidasa 1 (TPP1) y porcentajes de actividad determinadas en gotas de sangre seca en papel en padres de pacientes con diagnóstico clínico de CLN2
| Padres | Actividad de TPP1 nmol/mancha | % de actividad basado en el promedio | Estandarización al 100% |
| 1 (M) | 0,20 | 100 | 42,55 |
| 2 (F) | 0,07 | 35 | 14,9 |
| 3 | 0,17 | 85 | 36,17 |
| 4 | 0,06 | 30 | 12,76 |
| 5 | 0,18 | 90 | 38,29 |
| 6 | 0,28 | 140 | 59,57 |
| 7 | 0,47 | 235 | 100 |
| 8 | 0,36 | 180 | 76,6 |
| 9 | 0,27 | 135 | 57,45 |
| 10 | 0,08 | 40 | 17,02 |
| 11 | 0,27 | 135 | 57,45 |
| 12 | 0,12 | 60 | 25,53 |
| 13 | 0,33 | 165 | 70,2 |
| 14 | 0,41 | 205 | 87,23 |
Controles: n=36.
Promedio de actividad en adultos sanos: 0,20 nmol/mancha±0,08.
Rango de actividad enzimática: 0,11-0,5 nmol/mancha.
Métodos para el análisis enzimático basados en sustratos fluorescentes han sido desarrollados usando varios tejidos y fluidos biológicos que incluyen leucocitos plasmáticos y fibroblastos para el diagnóstico de las ceroidolipofuscinosis13. Sin embargo, el poder medir la actividad enzimática en muestras de sangre seca en papel calibrado permite el traslado de muestras y conservación a temperatura de ambiente de sitios lejanos a laboratorios especiales, pudiendo mantener su actividad enzimática hasta por 2 semanas12.
La determinación de la actividad enzimática de TPP1 en fibroblastos y leucocitos de pacientes con CLN2 y controles sanos por espectrofluorometría2,14-19 ha permitido establecer el diagnostico enzimático y decidir la secuenciación del gen para el diagnóstico molecular de CLN2.
Chamoles et al. (2001) inician el análisis enzimático lisosomal en gotas de sangre seca sobre papel como una alternativa a las técnicas tradicionales (espectrofluorometría) basado en programas de cribado neonatal. Ellos observaron que la actividad enzimática para hexoxaminidasas y ß-galactosidasa (en muestras de sangre seca en papel de filtro) se conservaban a pesar de estar almacenadas por algunas semanas a temperatura ambiente20.
En nuestro trabajo, se establecieron los valores referenciales de actividad de la enzima TPP1 en niños y adultos sanos y de ambos sexos en población venezolana. El rango de referencia en niños sanos en edad preescolar para la actividad de la enzima TPP1 fue de 0,11-0,45 nmol/mancha con promedio de 0,17±0,05 nmol/mancha. Asimismo el rango de referencia en adultos sanos fue de 0,11-0,50 nmol/mancha, siendo su promedio de 0,20±0,08 nmol/mancha. En el trabajo presentado por Zoltan et al.12 (2003) los valores de referencia encontrados para la población normal oscilan entre 0,1-0,67 nmol/mancha12. En el estudio realizado por Zoltan et al.12 (2003) y en el nuestro los valores de referencia para la población normal son similares.
El promedio de la actividad enzimática en nuestros pacientes fue de 0,03 nmol/mancha (tabla 1), mientras que los reportados por Zoltan et al.12 (2003) exhibían una actividad nula.
La edad de inicio de la enfermedad en los pacientes con CLN2 fue > 2 años, lo cual concuerda con la edad reportada por otros estudios2,17-19,21.
El desarrollo de esta metodología permite el diagnóstico enzimático de la CLN2 en forma rápida, sencilla y económica, y a su vez, el traslado de las muestras a temperatura de ambiente a laboratorios especializados, conservando su actividad enzimática. Además, el diagnóstico enzimático permite decidir ir a la etapa de análisis molecular del gen de la TPP1.
Este trabajo representa el primer estudio en Venezuela basado en un sustrato fluorogénico para establecer el diagnóstico enzimático de ceroidolipofuscinosis infantil tardía (CLN2) en nuestra población y el segundo en Latinoamérica.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
