



Los trastornos congénitos de la glucosilación constituyen un grupo de situaciones originadas por un defecto en la síntesis de las glucoproteínas. Sus manifestaciones pueden afectar a diversos órganos.
ObjetivosDar a conocer dos nuevos pacientes afectados de esta patología para contribuir a difundir el conocimiento de esta entidad.
MétodosPresentamos 2 pacientes con las manifestaciones clínicas, radiológicas, analíticas y genéticas compatibles con CD.
ConclusionesLos trastornos de la glucosilación constituyen un grupo de situaciones que se deben tener en cuenta en el diagnóstico de un paciente con un cuadro neurológico de origen inexplicable, en particular si asocia alteraciones hepáticas o de la coagulación.
Congenital glycosylation disorders (CGDs) are a group of disorders caused by a defect in glycoprotein synthesis. Clinical manifestations may affect to different organs.
AimsTo describe two new patients cases with a CGD in order to make paediatricians aware of this disorder.
Clinical casesTwo new cases of different age and gender are presented, showing clinical manifestations, and radiological and laboratory findings compatible with CGD. One of the cases was followed up for several years.
ConclusionsGlycosylation disorders are a group of conditions to bear in mind when considering the diagnosis of a patient with neurological symptoms of unexplained origin, particularly in those cases that include a delay in psychomotor activity, low muscle tone, epilepsy, and hepatic or coagulation disorders, as well as in patients with cerebellar or olivopontocerebellar atrophy.
Los trastornos congénitos de la glucosilación de las proteínas constituyen una nueva patología originada por un defecto en la síntesis de las glicoproteínas. La variante mejor conocida es la descrita por Norman1, para la que en los últimos años del pasado siglo2 se aportaron las bases del error metabólico responsable. Recientemente se ha propuesto cambiar su denominación por otra con el símbolo del gen seguido por -CDG3.
Dado el reducido número de pacientes descritos, aportamos 2 sujetos más para difundir su conocimiento.
Casos clínicosCaso 1Niña, hija única de padres sanos, no consanguíneos, sin antecedentes familiares reseñables. Embarazo controlado, parto: cesárea a las 37,4 semanas, por presentación podálica. Apgar 9/10. Peso: 2,590 g (percentil 25-50), talla: 46cm (percentil 25), perímetro cefálico: 33,5cm (percentil 50-75). Lactancia mixta desde el nacimiento. Inicio de ictericia al segundo día, alcanzando bilirrubina directa de 0,6mg/dl y total de 16,9mg/dl (sometida a fototerapia); la ictericia se prolonga el primer mes sin que la bilirrubina total sobrepase los 16mg/dl. A los 3 meses se observa un fallo del crecimiento, ausencia de sostén cefálico y de fijación de la mirada; los estudios de ecografía cerebral, otoemisiones y potenciales visuales fueron normales, iniciándose apoyos de fisioterapia y estimulación.
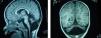
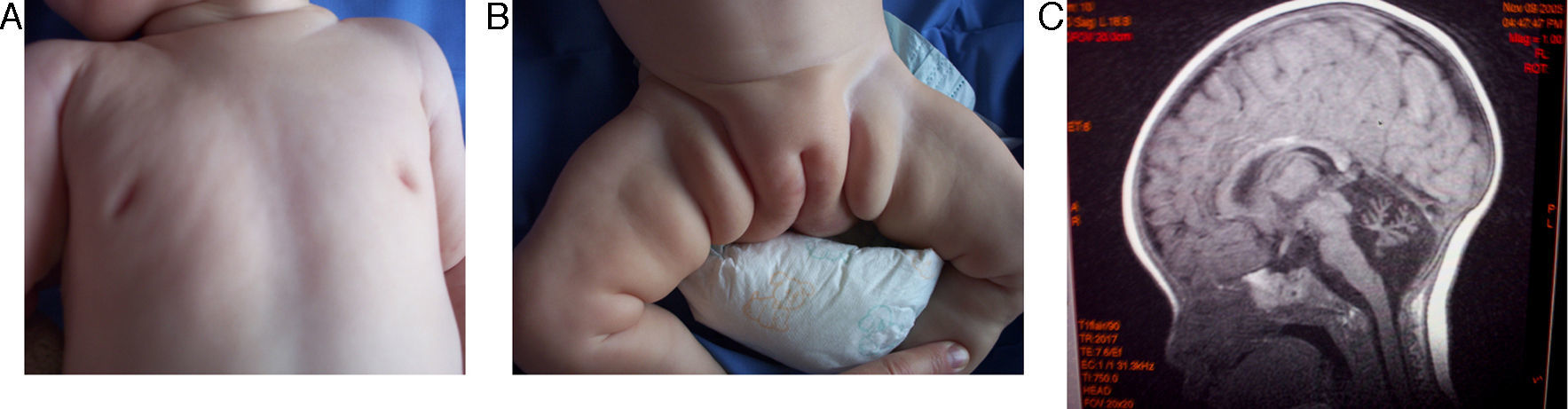
A los 6 meses y medio consulta por vómitos y diarrea. Exploración: dolicocefalia, dismorfia craneal (frente amplia, raíz nasal ancha, hendiduras palpebrales dirigidas hacia arriba y fuera); hipotonía y reflejos normales. No sostén cefálico, no sigue con la mirada, ni sonríe. Deformidad torácica en quilla y mamilas invertidas (fig. 1A). Hígado palpable 2cm, hipertrofia de labios mayores, lipodistrofia glútea (fig. 1B). Peso y talla por debajo del percentil 3 y perímetro cefálico en percentil 10. Laboratorio: elevación e inversión de GOT y GPT; coagulación, pH y gases, cariotipo, alfa 1 antitripsina: normales. Electroencefalograma y velocidad de conducción nerviosa: normales. Resonancia encefálica: atrofia cerebelosa (fig. 1C).
Porcentaje de transferrina deficiente en carbohidratos: 38% (muy elevada) y el patrón de sialotransferrinas por isoelectroenfoque mostraba presencia de asialo y monosialotrasferrina y marcada presencia de disialotransferrina. Genética: la paciente mostró las mutaciones D65Y (c.193G>T) (también presente en el padre, un tío y abuelo paterno) y una segunda mutación IVS 7nt-9T>G (c640-95>G) (encontrada en la madre y el abuelo materno).
Caso 2Varón de 13 años, segundo de 2 hijos (hermano sano). Antecedentes familiares: sin interés. Embarazo normal; parto a término, cefálico, eutócico. Peso neonatal: 2.970 g. Periodo neonatal: normal.
Retraso psicomotor: sostén cefálico a los 5 meses, sedestación a los 3 años; no ha alcanzado deambulación autónoma. Exploración: estrabismo convergente de ojo izquierdo, hipotonía muscular con hiporreflexia, escoliosis, sólo emite bisílabos. Peso, talla, perímetro cefálico: inferiores al percentil 3. Laboratorio: hematíes, leucocitos, plaquetas, amonio, ácido láctico, ácidos orgánicos, aminoácidos, coagulación: normales. Electroencefalograma, electromiograma, velocidad de conducción nerviosa, electrocardiograma: normales. Resonancia encefálica: atrofia cerebelosa (fig. 2). La transferrina sérica presentó valores de 256mg/dl (referencia: 200-360) con elevación de la disialotransferrina (10,9% de la transferrina total siendo los valores de referencia inferiores al 2,6%). Estudio genético: el paciente presenta dos mutaciones en el gen PMM2 -mutación p.V44A (c.131T>C) y p.R141H (c.422G>A)-; el padre y hermano presentan la mutación p.R141H y la madre la p.V44A.
Comentarios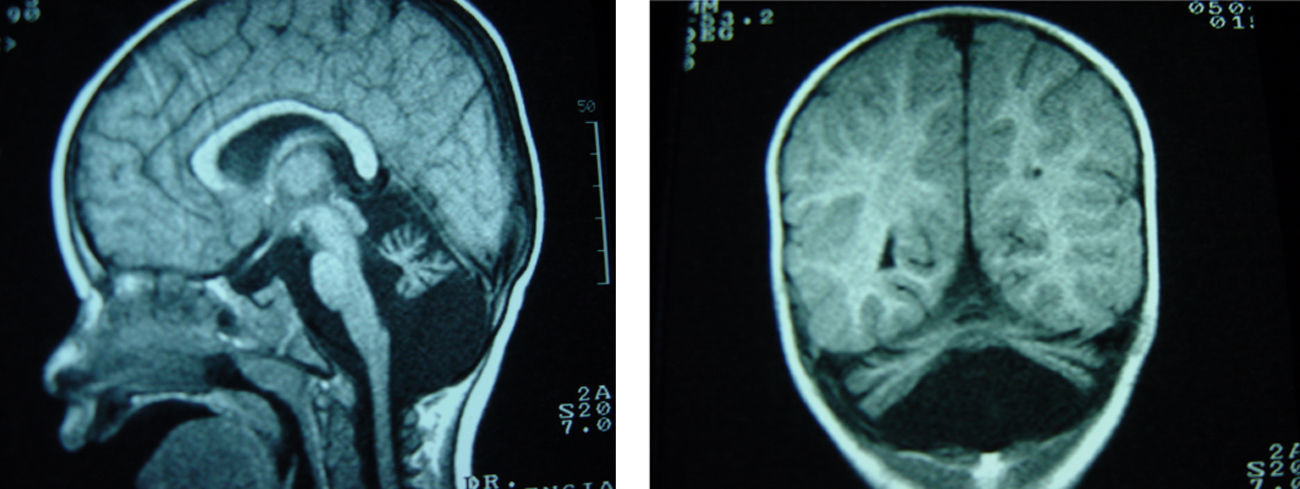
Los pacientes inicialmente descritos con CDG mostraban transferrina anormal (aumento de di y asialotransferrina); se denominaron CDG-Ia. Más tarde se describieron otros con patrones de transferrina distintos: CDG-IIa4–6.
El tipo más frecuente es CDG-Ia (síndrome de Jaeken), originado por mutaciones en el gen PMM2 (phosphomannomutase-2) localizado en 16p13.27; codifica la fosfomanomutasa.
Su clínica evoluciona en varias etapas: fase infantil con dismorfias múltiples, mamilas invertidas, anormal distribución de la grasa (evidente en el paciente 1), criptorquidia, infecciones de repetición, cardiomiopatía, hipotiroidismo, nefropatía, vómitos, diarrea, coagulopatía, hepatopatía y alteraciones neurológicas8. En esta fase fallecen en torno del 20-25% de los pacientes con CDG Ia.
La segunda fase cursa con crisis epilépticas, facilitadas por infecciones; la tercera fase cursa con ataxia, debilidad muscular, lenguaje pobre y retinopatía pigmentaria. Los supervivientes presentan discreto retraso con ataxia severa, hipogonadismo, con o sin deformidades torácicas y de la columna (evidentes en el paciente 2), con afectación del desarrollo puberal en las mujeres (puede ser normal)9.
La radiología muestra atrofia cerebelosa10 y del tronco cerebral (fosa posterior «casi vacía»), como los dos pacientes que comentamos.
Salvo para el CDG-Ib (administración de manosa) y el CDG-IIc (suplemento de fucosa oral), no disponemos de tratamiento eficaz; la atención a estos pacientes se basa en medidas sintomáticas: fisioterapia, estimulación, atención a los problemas oculares, de nutrición, etc.
Para algunos autores11,12 la ataxia con atrofia de la capa de granos de Norman y la enfermedad de Jaeken constituyen una misma entidad (enfermedad de Norman-Jaeken).
En resumen, dada la heterogeneidad clínica de estos enfermos se debe sospechar un CDG —y realizar un estudio de la glucosilación de la transferrina— en todo paciente con un cuadro neurológico inexplicable, en particular si junto a retraso psicomotor, hipotonía y epilepsia asocia alteraciones hepáticas o de la coagulación, así como en casos de hipoplasia cerebelosa u olivo pontocerebelosa de aparición en el período neonatal; en los pacientes que presentan una ataxia hereditaria aparentemente recesiva con hipoplasia de cerebelo es recomendable determinar la tasa de fosfomanomutasa cuando la transferrina es normal o no concluyente13.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A la Dra. Matíes, del Hospital Ramón y Cajal, el estudio de asialotrasnferrina (caso 1), y a la Prof. M. Ugarte, del Centro de Diagnóstico de Enfermedades Moleculares, la genética de ambos.
