



La fiebre mediterránea familiar (FMF) es una enfermedad hereditaria que se caracteriza por episodios breves y recurrentes de fiebre y dolor por inflamación de una o varias serosas (peritoneo, pleura, pericardio, sinovial o túnica vaginal del testículo). La amiloidosis es su complicación más importante y suele ser la principal causa de muerte en los casos en que se presenta. El diagnóstico se basa en la clínica y se confirma mediante pruebas genéticas. Para el tratamiento, se utiliza colchicina a 0,02-0,03mg/kg/día, que permite tanto evitar la crisis como el desarrollo de la insuficiencia la renal. Presentamos el caso de un niño de 13 años en el que se diagnosticó FMF tras varios episodios coincidentes con fiebre de pericarditis con taponamiento cardiaco. La confirmación genética mostró un patrón de herencia poco frecuente autosómico dominante.
Familial Mediterranean fever (FMF) is a hereditary disease characterized by brief, recurring and self-limited episodes of fever and pain with inflammation, of one or several serous (peritoneum, pleura, pericardium, synovial or vaginal tunic of the testicle). Amyloidosis is its more important complication and the principal reason of death in the cases in which it appears. Diagnosis is based on the clinic and is confirmed by genetic tests. The treatment with Colchicine (0,02-0,03mg/kg/day) prevents the recurrence of FMF attacks and the development of secondary (AA) amyloidosis. We report a case of a 13-year-old child in which FMF was diagnosed after several coincidental episodes with fever, pericarditis and cardiac tamponade. The genetic confirmation showed an autosomal dominant inheritance that is less frecuent than the recesive form, in this disease.
La FMF es una entidad poco frecuente que queda englobada en las fiebres periódicas. Tiene un componente hereditario fundamental, siendo una enfermedad autosómica recesiva en la mayoría de los casos y que se caracteriza por episodios recurrentes, breves y autolimitados de fiebre con poliserositis, que recidivan a intervalos regulares. La complicación más importante es la amiloidosis, que puede desarrollar insuficiencia renal, siendo esta la causa de muerte en los casos en que se presenta1.
La afección genética se localiza en un intervalo pequeño del brazo corto del cromosoma 16.p13.3. El gen se denomina MEFV, mediante el que se codifica una proteína llamada pirina o maresnostrina. Las mutaciones más frecuentes son M694V, V726A, M694I, M680I, E148Q 2. La transmisión de la FMF ha sido definida clásicamente como autosómica recesiva, aunque el avance en las técnicas de diagnóstico genético ha mostrado diversos patrones de herencia, siendo algunos de ellos autosómicos dominantes3. La incidencia es mayor en la población de origen mediterráneo, fundamentalmente en poblaciones como judíos sefardíes, turcos y armenios, y de ascendencia árabe, siendo menos frecuente en griegos, italianos o españoles.
La patogenia exacta en los procesos agudos de la FMF es desconocida. Se especula con que la piridina mutada que hay en estos pacientes causa una apoptosis defectuosa y una estimulación del procesamiento y secreción de interleucina 1 (IL-1), que es responsable de una inflamación descontrolada con gran variedad de citocinas, entre ellas la enzima que genera el factor C5a. Todos estos factores inflamatorios son los causantes de las crisis en la FMF4.
Las manifestaciones clínicas suelen aparecer antes de los 5 años. Los episodios agudos duran de 1 a 4 días y se caracterizan por fiebre y uno o más de otros síntomas. El más frecuente es la peritonitis (90%), manifestado por dolor abdominal, artritis o artralgias (85%), y pleuritis (20%) en forma de dolor torácico5,6. Más rara es la afectación de otros tejidos serosos, como la túnica vaginal testicular7. Otras afecciones descritas son hipoalderosterolismo8, mialgias, erisipela9, afectación neurológica10 o púrpura de Schönlein-Henoch. La pericarditis es un síntoma infrecuente, aunque bien conocido de la enfermedad11,12.
El diagnóstico de presunción se basa en la clínica y se confirma mediante pruebas genéticas. El tratamiento consiste en evitar la aparición de las crisis, mediante tratamiento profiláctico con colchicina a 0,02-0,03mg/kg/día, que permite tanto evitar la crisis como que se desarrolle insuficiencia renal por amiloidosis13,14.
Caso clínicoPresentamos el caso de un niño de 13 años de edad, que acude a urgencias por fiebre y dolor torácico continuo, opresivo, de 24 h de evolución, que empeoraba con el decúbito supino y la tos. No presentaba dificultad respiratoria. Las constantes eran normales. La exploración física mostraba una afectación del estado general, destacando unos tonos cardiacos atenuados y hepatomegalia de 3cm, sin signos de regurgitación yugular.
Durante el último año, había presentado varios episodios de dolor torácico coincidente con picos febriles que había cedido en pocos días con tratamiento con ibuprofeno. No tenía enfermedades de base. El padre refería que, en una ocasión, había presentado un cuadro de pericarditis autolimitada sin que se encontrase causa aparente, y el tío paterno refería dolores articulares en la infancia. No había enfermedades hereditarias conocidas en la familia.
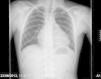
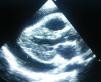
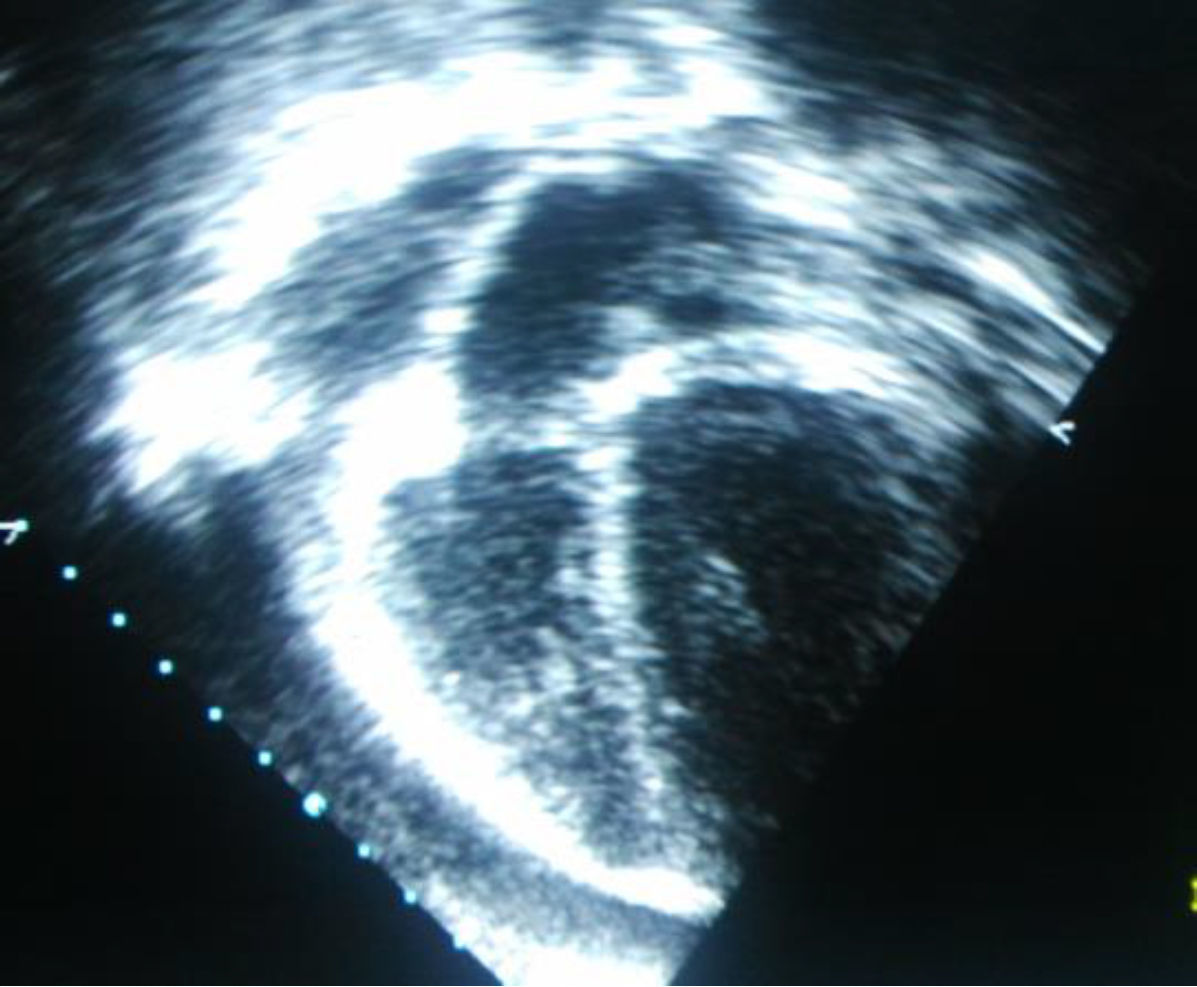
Se realizó una radiografía de tórax (fig. 1) que objetivaba cardiomegalia, un electrocardiograma donde observamos un leve aumento de ST con voltajes disminuidos, y una analítica que presentaba leucocitosis con predominio de neutrófilos y aumento de reactantes de fase aguda con proteína C reactiva de 10mg/dl. La fracción MB de creatincinasa y la troponina i eran negativas. La ecocardiografía (figs. 2 y 3) muestra un derrame pericárdico moderado de 18mm en la aurícula derecha (AD), sin signos en este momento de taponamiento cardiaco.
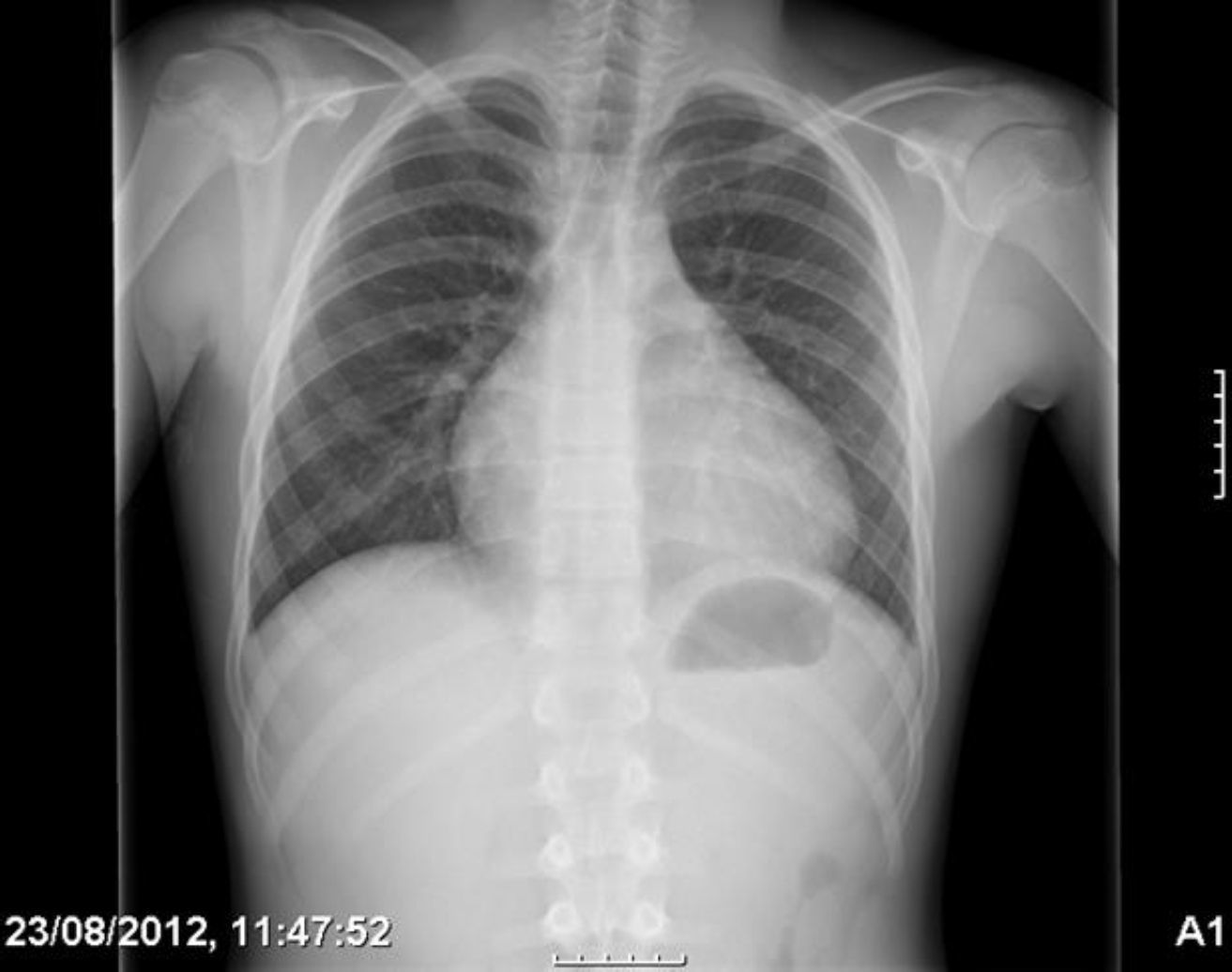
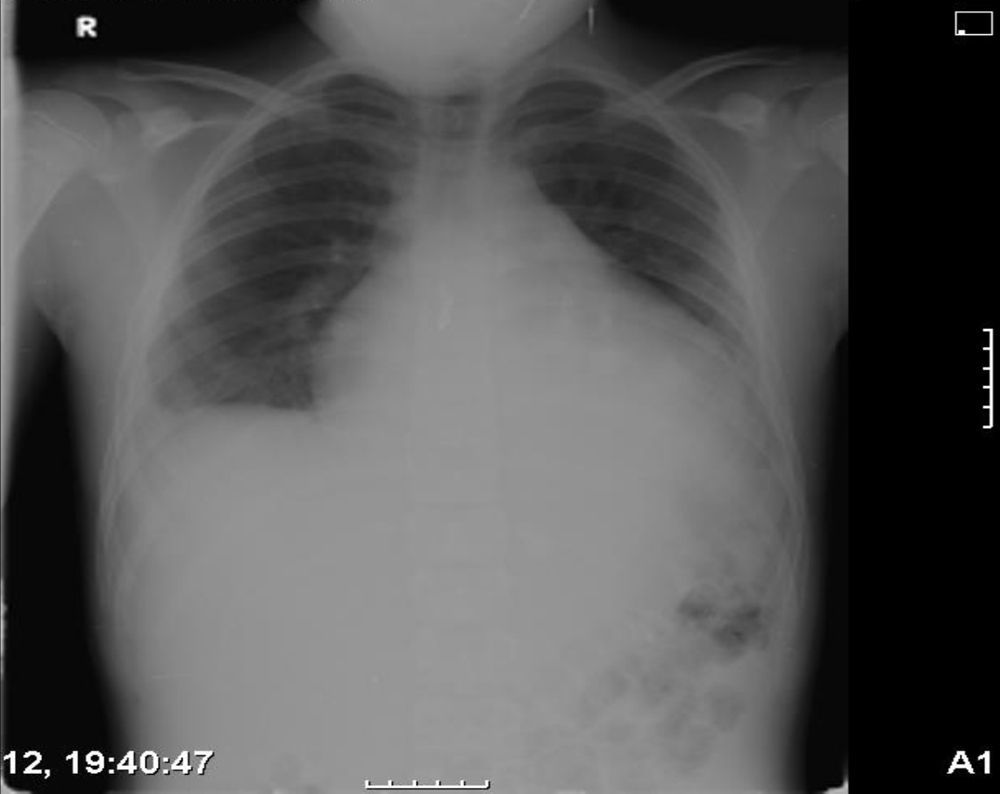
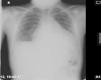
Se ingresa con tratamiento con antibióticos e indometacina por vía intravenosa. A las 30 h de ingreso, se produce un empeoramiento clínico con polipnea, tos frecuente y aumento del trabajo respiratorio. La radiografía de tórax de control (fig. 2) presentaba cardiomegalia y derrame pleural bilateral. En la ecocardiografía (fig. 4) muestra un aumento de derrame pericárdico (26mm) en AD, con signos de colapso de AD y ventrículo derecho en diástole, aumento de tamaño de la vena cava inferior. En la ecografía abdominal se observan signos de congestió hepática.
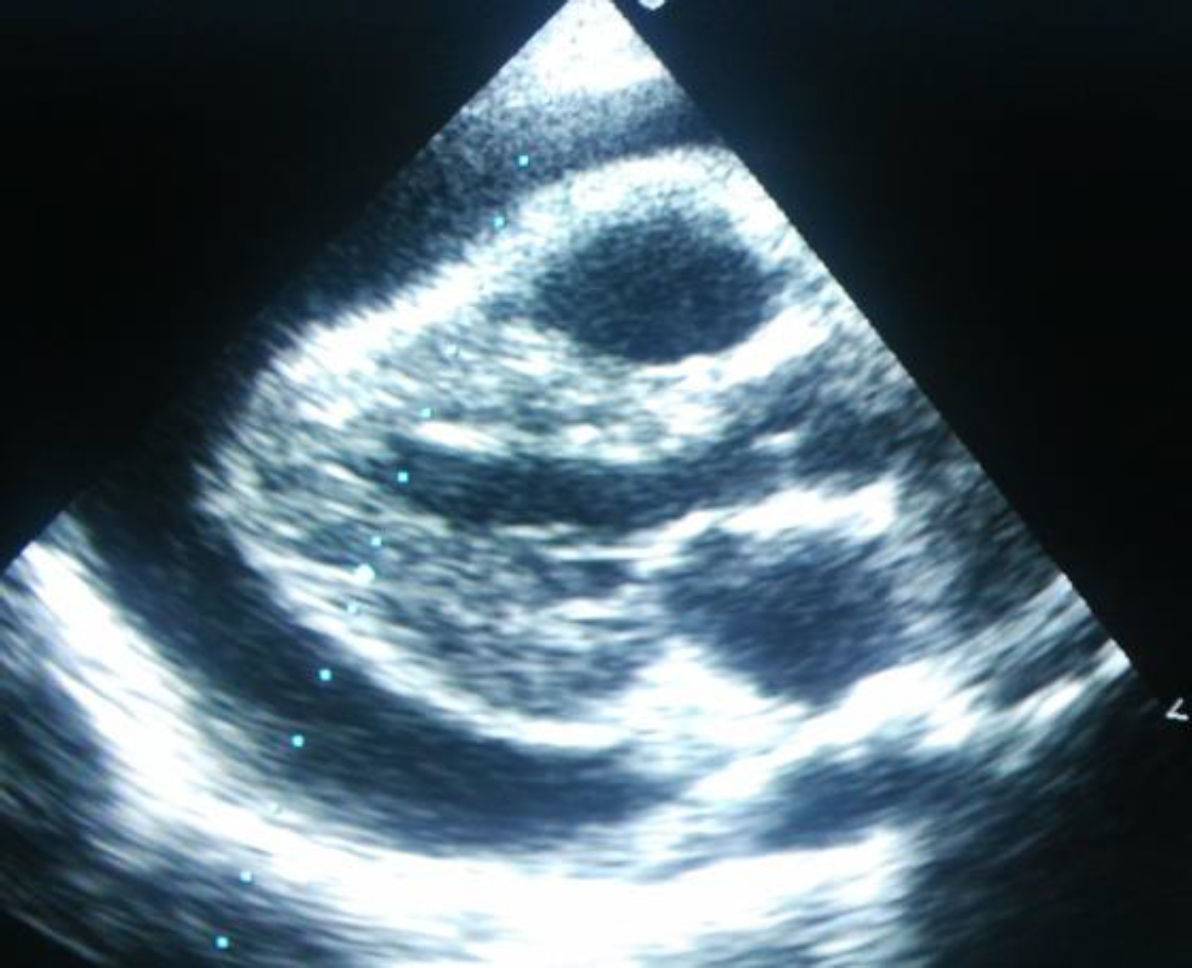
Ante la situación de gravedad, se traslada a la UCI pediátrica, donde se realiza pericardiocentesis evacuadora urgente, extrayendo 700ml de líquido seroso, solicitándose serología y cultivo del mismo, siendo ambos negativos. Se inicia tratamiento con indometacina, corticoides y diuréticos por vía intravenosa, con mejoría progresiva.
Pasado un mes del episodio, repite un cuadro similar con fiebre, dolor torácico y refiriendo dolor articular en los miembros inferiores durante el mes previo. Se objetiva derrame pericárdico severo, precisando pericardiocentesis y tratamiento con indometacina, corticoides por vía intravenosa y colchicina por vía oral, con mejoría del cuadro y alta sin tratamiento de mantenimiento.
A los 2 meses del episodio inicial, reingresa con la misma clínica y derrame pericárdico leve-moderado. Precisa mismo tratamiento médico agudo, dejando la colchicina como tratamiento de mantenimiento a 0,03mg/kg/día.
Ante la situación de gravedad, se traslada a la UCI pediátrica, donde se realiza pericardiocentesis evacuadora urgente, extrayendo 700ml de líquido seroso, solicitándose serología y cultivo del mismo, siendo ambos negativos. Se inicia tratamiento con indometacina, corticoides y diuréticos por vía intravenosa, con mejoría progresiva.
Las analíticas en periodos intercrisis mostraban velocidad de sedimentación, complementos normales, anticuerpos antinucleares y anti-ADN negativos, antígenos leucocitarios humanos B27 negativos, hormonas tiroideas normales y factor reumatoide negativo e inmunoglobulinas normales. Analíticas de orina sin afectación renal.
Dado los episodios repetidos pericarditis, y teniendo en cuenta los antecedentes familiares descritos, se sospecha FMF, confirmándose mediante estudio genético al detectarse la presencia de la variante genética p.Glu-148-Gln (p. E148Q), localizada en el exón 2 y en solo uno de los alelos del gen (heterocigosis)15,16.
En los últimos meses, ha presentado 2 episodios que han sido tratados con terapia biológica con IL-1RA (Anakinra®)17, con buenos resultados, desapareciendo la clínica en pocas horas. Actualmente, continúa tratamiento de mantenimiento con colchicina.
DiscusiónLa FMF es una entidad poco frecuente en nuestro medio; debemos conocerla puesto que el 90% de los casos comienza antes de los 20 años. La forma de presentación típica son episodios recurrentes de dolor abdominal, artritis o artralgias durante 1-4 días, coincidiendo con fiebre. No por ello debemos olvidar que puede presentarse con otra sintomatología, como en nuestro caso, que es atípico por presentar pericarditis repetidas en proceso febril como única sintomatología, sin tener las otras afectaciones comunes en la FMF, si bien esta forma de presentación ha sido descrita previamente18. Si se emplea ecografía para el diagnóstico de pericarditis en las crisis de FMF, se ha descrito una incidencia de hasta el 27% de los casos estudiados11, incluso hay publicaciones donde es referida la pericarditis recurrente en niños entre 8 y 11 años19. En nuestro paciente, la forma de presentación inicial con su llegada a urgencias, con un derrame pericárdico severo que precisó pericardiocentesis, ha de llamar la atención sobre la posibilidad de eventos potencialmente graves.
El precoz y correcto tratamiento es vital para evitar las crisis y sobre todo la amiloidosis AA, que puede degenerar en insuficiencia renal. La colchicina se ha mostrado eficaz sin apenas efectos secundarios para el tratamiento de por vida de esta afección.
Los antecedentes familiares son fundamentales para el diagnóstico de sospecha, ya que suele haber casos de FMF en la familia. En nuestro caso, no estaban diagnosticados de dicha enfermedad, si bien los síntomas leves de un episodio de pericarditis en el padre y artritis en el tío paterno ayudaron en el diagnóstico de presunción.
La genética encontrada es peculiar, puesto que la FMF clásicamente se definía como enfermedad autosómica recesiva, pero en nuestro caso, al igual que otras formas descritas para la FMF, es autosómica dominante15,16. La mutación E148Q es una de las descritas como frecuentes. Específicamente, la mutación de nuestro caso Glu 148-Gln ha sido mencionada como una mutación recesiva con una penetrancia reducida15.
La FMF en una afección que requiere una sospecha diagnóstica alta; los síntomas clínicos típicos son muy variables o únicos y atípicos, como en nuestro caso. Los antecedentes familiares, tanto de enfermedades conocidas de como síntomas aislados, deben hacernos sospechar, pese a que no exista un diagnóstico de la enfermedad.
El tratamiento con colchicina de por vida parece claro para evitar los brotes, así como el desarrollo de la enfermedad hacia amiloidosis y fallo renal. Sin tratamiento, entre un 33 y un 50% de los niños y hasta el 75% de los adultos producen amiloidosis AA. Se debe al depósito lento en órganos y tejidos de la sustancia amiloide, procedente de la proteína sérica del amiloide (SAA-1). La mortalidad de la FMF suele deberse a complicaciones de la insuficiencia renal, así como a la infección, el tromboembolismo y la uremia. Recientemente, la utilización de IL-1RA biológico (Anakinra®), en aquellos casos de fallo o intolerancia a la colchicina, ha mostrado resultados esperanzadores17.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Al Dr. Eliseo Pascual, del Servicio de Reumatología del Hospital General de Alicante.
