


Sr. Editor:
El síndrome de Edwards es un síndrome polimalformativo debido a la existencia de tres cromosomas 18. Su incidencia se estima entre 1/3.000-1/6.000 recién nacidos vivos, aunque su incidencia real depende del porcentaje de diagnóstico prenatal mediante la realización o no de amniocentesis 1. Es más frecuente en madres de edad avanzada, y llega incluso a 1/500 a la edad de 43 años. El riesgo de recurrencia se estima en el 0,55 % 2. El 95 % de los casos corresponden a trisomía completa por no disyunción, siendo el resto trisomía por translocación. La trisomía parcial y el mosaicismo para la trisomía 18 suelen presentar un fenotipo incompleto 1. Aparece con mayor frecuencia en mujeres, y suelen presentar numerosas anomalías congénitas 1,3. Las más frecuentes son el retraso del crecimiento intrauterino, las anomalías craneofaciales, en extremidades, esternón corto, malformaciones urogenitales, gastrointestinales, del sistema nervioso central (SNC), y, especialmente, cardiopatías congénitas (90 %) 1,4. El 90-95 % mueren ya en el primer año de vida 1,5-7, siendo excepcionales los casos que llegan a la adolescencia 7.
Se presenta un caso de trisomía 18 de supervivencia prolongada. Se trataba de un varón, hijo de madre de 43 años de edad, con dos abortos previos, y padre de 49 años, nacido a término por cesárea tras embarazo controlado, sin realización de amniocentesis por decisión familiar, que mostraba depresión neonatal (Apgar de 2/5/8), peso 1.840 g (< P10), longitud 44 cm (< P10), y perímetro craneal 32 cm (< P10). Presentaba rasgos dismórficos múltiples, con occipucio prominente, micrognatia, blefarofimosis, pabellones auriculares malformados y de implantación baja, sindactilias en pies, clinodactilias y tendencia a puño cerrado en manos, luxación de caderas, criptorquidia, hipotonía generalizada, soplo sistólico y posteriormente hernia inguinal bilateral. El cariotipo demostró en todas las metafases estudiadas una trisomía 18 (47, XY, + 18). Sucesivamente se practicaron otros exámenes complementarios, entre los que destacaban los siguientes. En la radiología de tórax se observó cardiomegalia y costillas delgadas. La ecocardiografía mostró doble cámara de salida de ventrículo derecho con comunicación interventricular subaórtica e hipertensión pulmonar grave, decidiendo una actitud no quirúrgica. La ecografía cerebral fue normal, y en la abdominal se demostró una hidronefrosis bilateral y riñón en herradura.
Ha presentado pielonefritis, gastroenteritis y neumonías de repetición, y numerosas crisis convulsivas tónicas (con puntas y polipunta-ondas generalizadas en el EEG). Por su cardiopatía presentó cianosis progresiva, con crisis hipoxémicas de frecuencia creciente y poliglobulia intensa (hematócrito 63,8 % en último control), que precisó en los últimos años oxigenoterapia domiciliaria. Asimismo, realiza rehabilitación por cifoescoliosis intensa (fig. 1). A pesar de todo, el niño ha mantenido siempre alimentación oral, administrada por otra persona, con un estado nutricional aceptable, una higiene escrupulosa corporal y bucodental, calendario vacunal correcto, y fisioterapia frecuente domiciliaria, con medidas ortopédicas posturales, que han permitido su transporte habitual y cierta autonomía hasta el final. No obstante, el retraso psicomotor era profundo, sin lenguaje ni manifestaciones emocionales, excepto la expresión o no de dolor, con hipertonía generalizada. A los 14 años y un mes presentó parada cardiorrespiratoria y muerte en el contexto de un proceso bronconeumónico.
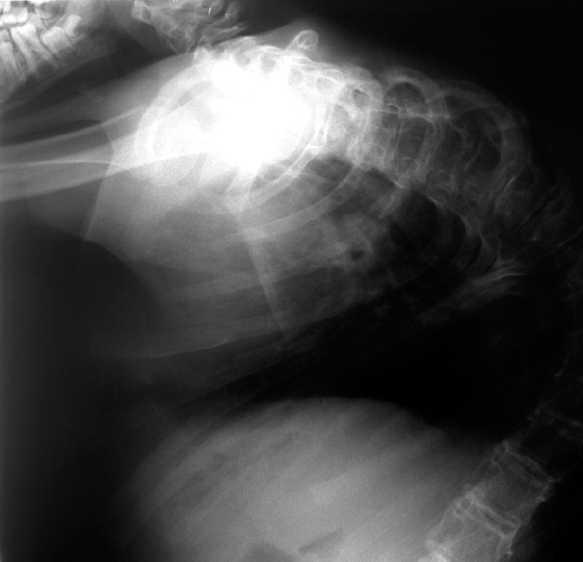
Figura 1. Radiografía de tórax del enfermo, a la edad de 14 años.
El síndrome de Edwards presenta una supervivencia media inferior a un mes 3,5-9, y casi todos los pacientes fallecen antes del año 1,5-7, sin que este dato se haya modificado demasiado con el paso del tiempo 8. Las primeras causas de defunción no son sorprendentemente las cardiopatías congénitas, presentes en casi todos los casos, sino la apnea central o las neumonías 4,8. Aunque el tema es controvertido 7, para algunos autores el tratamiento quirúrgico en cardiopatías graves no estaría justificado, dadas las características del paciente, y porque esta actitud conservadora no pareció acortar su vida 3,5, como probablemente ocurrió en nuestro enfermo.
No se conocen exactamente los factores que contribuyen a la supervivencia prolongada en esos casos raros de mayor longevidad. Sólo se ha descrito que el sexo femenino y, en general, los niños de razas distintas a la blanca, tendrían una mayor tasa de supervivencia 5. Aunque parece razonable pensar que en los casos de mosaicismo la supervivencia sea mayor, esto no se ha demostrado 2. En cualquier caso, ninguno de esos hechos se dieron en nuestro paciente. Sin embargo, sí fueron muy patentes factores como el grado de cuidado y dedicación al niño, aspectos psicológicos y de apoyo social, que probablemente sí podrían influir en su longevidad 10. El tratamiento clínico del niño depende de las expectativas de vida, en términos de supervivencia y calidad de vida. Los que superan el año de vida suelen presentar dificultades en la alimentación, y con frecuencia precisan técnicas artificiales para la misma, escoliosis progresiva, estreñimiento persistente, infecciones de repetición y, en todos los casos, importantes limitaciones psicomotoras 1. No obstante, en la toma de decisiones y consejo paterno es importante conocer también estos infrecuentes casos de larga supervivencia.
