





Niña de 4 años remitida a cardiología infantil por sospecha de colagenopatía (hiperlaxitud, hipotonía y rasgos dismórficos) con elongación del arco aórtico radiológico.
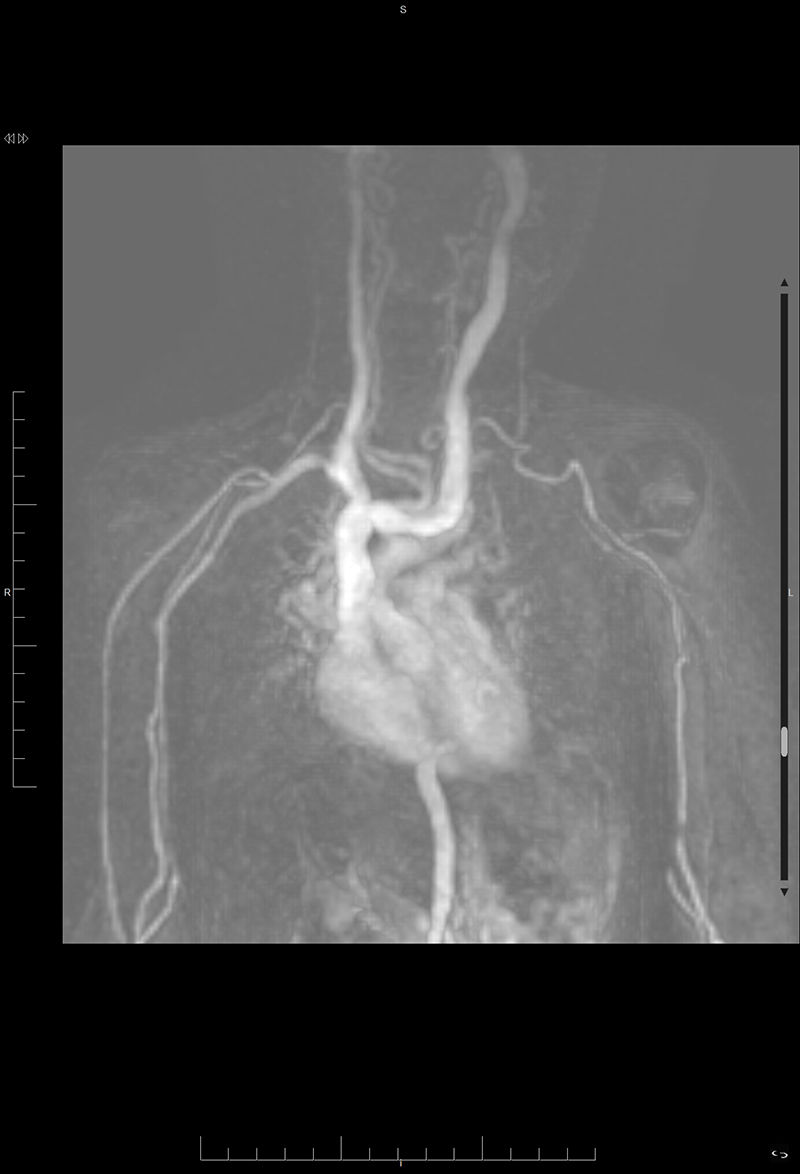
La ecocardiografía destaca un arco aórtico rectangular y troncos supraaórticos (TSA) elongados. La angio-resonancia magnética (angio-RM) evidencia una severa elongación de la aorta torácica y los TSA con hipoplasia de cayado distal. Bucles en las transiciones: ascendente-cayado, cayado-descendente y torácica-abdominal (fig. 1). La aorta se dirige hacia posterior enmarcando el corazón, cruzando al lado derecho en zigzag a la altura del diafragma comprimiendo la vena cava inferior. Llamativa elongación del tronco celíaco, mesentérica superior y renales (fig. 2) como de arterias vertebrales, subclavias, axilares y braquiales (fig. 3), y bucles vasculares cerebrales.

Angio-RMN. Reconstrucción volumen rendering en 3 dimensiones de aorta torácica y troncos supraaórticos. Severa elongación de aorta torácica con hipoplasia de cayado distal. Bucles en la transición entre aorta ascendente y transversa, transversa y descendente, y en la transición toraco-abdominal. Troncos supraaórticos muy elongados.
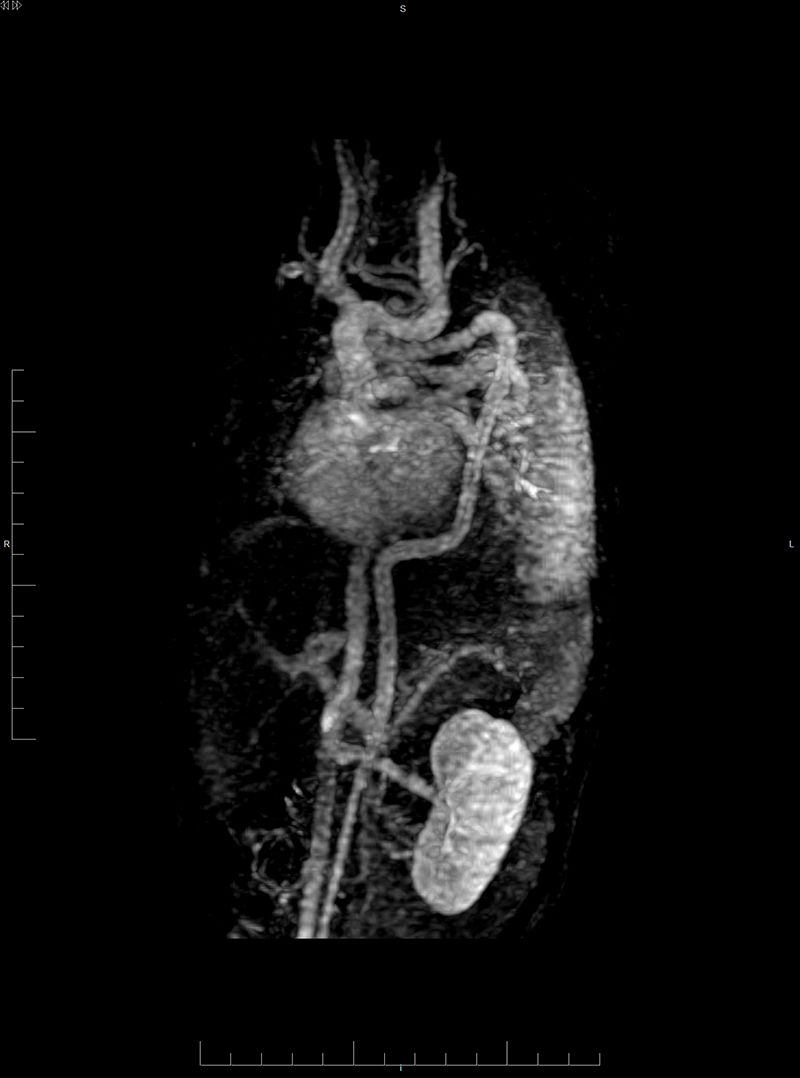
Angio-RMN. Reconstrucción MIP grueso que incluye los trayectos arteriales y venosos de tórax y abdomen. Severa elongación de aorta torácica con cayado distal hipoplásico. Marcadas angulación en aorta ascendente, al inicio de la aorta descendente y en la transición toraco-abdominal. Elongación de los troncos supraaórticos en «tirabuzón» y de las ramas viscerales de aorta abdominal.
El estudio genético identificó una variante patogénica descrita en homocigosis (delección c.1334delG, exón 3, gen SLC2A10) relacionada con el síndrome de tortuosidad arterial (STA).
El STA OMIM#208050 es una enfermedad rara autosómica recesiva del tejido conectivo, que se caracteriza por elongación, tortuosidad y predisposición de aneurismas en arterias de mediano y gran calibre, disección vascular y eventos isquémicos. Se asocia con estenosis de arterias pulmonares y/o aorta con o sin rasgos dismórficos, anomalías esqueléticas, oftálmicas e hipotonía. Hasta el momento se han descrito 23 mutaciones en SLC2A10 en unos 100 pacientes1.
Suele comenzar en la infancia temprana, con manifestaciones clínicas variables, dependiendo del territorio arterial implicado. El diagnóstico precoz y un seguimiento exhaustivo mediante pruebas de imagen y despistaje de anomalías asociadas tratará de evitar las complicaciones ofreciendo un mejor pronóstico a largo plazo2,3.
