






La forma clásica de hiperplasia suprarrenal congénita por déficit de 21-hidroxilasa (21-OHD) se debe a mutaciones del gen CYP21A2. La gran mayoría de los alelos deficientes muestran mutaciones que preexisten en un seudogen homólogo y localizado en tandem: CYP21A1. Los alelos se heredan de los padres portadores, y las mutaciones de novo en el transcurso de la gametogénesis o en el desarrollo fetal son excepcionales. Este artículo describe a una paciente afectada de 21-OHD clásico que presentó en su alelo materno la mutación de novo I172N en heterocigosis compuesta con la mutación grave R356W heredada del padre. La madre de la paciente resultó negativa en el estudio de mutaciones del gen CYP21A2. El estudio complementario de marcadores indirectos tipo microsatélite confirmó una segregación correcta de los alelos parentales. La mutación I172N (en heterocigosis compuesta con mutación nula) da lugar a un fenotipo muy característico neonatal virilizante que no asocia crisis de pérdida salina.
The classical form of congenital adrenal hyperplasia is the result of mutations in the 21-hydroxylase gene (CYP21A2). Most deficient alleles carry pre-existing mutations in the CYP21PA homologue pseudogene, located in tandem. Mutant alleles are inherited from carrier parents, and de novo mutations during gametogenesis or foetal development are exceptional. The present paper describes a de novo mutation occurring at the maternal allele (I172N) of a patient with a classical form of 21-hydroxylase deficiency, whose father was heterozygous for R356W. The mother did not carry the mutation. Microsatellite analyses confirmed a correct allelic segregation. The I172N mutation (in compound heterozygosity with a null mutation) gives rise to a virilizing phenotype not associated with salt-wasting.
La hiperplasia suprarrenal congénita (HSC) se debe en un 95% de los casos al déficit de la 21-hidroxilasa (21-OHD). Esta enfermedad autosómica recesiva presenta un amplio espectro de formas clínicas, desde las clásicas neonatales, que en su variante perdedora de sal pueden dañar la vida de estos niños, hasta formas atenuadas denominadas no clásicas, en que los síntomas aparecen en la etapa pediátrica o adulta, e incluso formas crípticas u oligosintomáticas.
Es una enfermedad común en sus formas leves, especialmente en el área mediterránea1, y no infrecuente en sus formas graves (frecuencia de portadores de mutación grave de 1:50–1:60), en las que cabe la posibilidad de plantear un tratamiento prenatal que previene la virilización de los fetos femeninos afectados2. Existe una estrecha correlación genotipo/fenotipo y se ha señalado que la genotipificación puede ser relevante para la categorización de estos pacientes3.
El cribado básico de 10 mutaciones puntuales (P30L, 655AoC→G, deleción 8pb, I172N, I236N/V237E/M239K, V281L, 306insT, Q318X, R356W y P453S), deleciones y conversiones permite caracterizar un gran número de alelos anormales. Existe un seudogen homólogo, duplicado en tándem, que presenta las alteraciones puntuales mencionadas y que, por procesos de recombinación asimétrica y conversión génica, puede originar los alelos anormales3–5. El porcentaje de alelos 21-OHD caracterizados tras el cribado básico de estas mutaciones recurrentes en nuestra población es del 93% para las formas clásicas y del 85% para las formas no clásicas (algo menor)6.
El pequeño porcentaje de alelos 21-OHD clásicos restante presenta mutaciones producidas por los mecanismos tradicionales, y su caracterización se realiza mediante la secuenciación completa del gen. Estas mutaciones no frecuentes o raras se encuentran generalmente en hemicigosis y heterocigosis compuesta con las mutaciones antes mencionadas5, aunque también se han descrito homocigotos, generalmente por consanguinidad7.
Actualmente, estas nuevas mutaciones se recogen en la base de datos HGMD (Human Gene Mutation Database) del Institute of Medical Genetics en Cardiff (http://www.hgmd.cf.ac.uk).
La frecuencia del déficit clásico, establecida en estudios retrospectivos en diferentes países8 a partir de los casos diagnosticados, osciló entre 1/15.000 y 1/23.044 nacimientos. La incidencia mundial de la HSC por 21-OHD determinada por los programas de cribado es del orden de 1/14.554 nacidos vivos, de los que aproximadamente el 75% pertenece al fenotipo con pérdida salina9. La American Academy of Pediatrics estima que la prevalencia de las formas clásicas de HSC por 21-OHD se encuentra aproximadamente entre 1/12.000 y 1/15.000 nacimientos9.
En varios países de nuestro entorno y en algunas comunidades autónomas del ámbito nacional existen centros que incluyen la detección neonatal de la HSC para su área de influencia sanitaria; Extremadura, Madrid, Castilla-La Mancha, Galicia y Aragón lo incluyen en la actualidad, lo que representa una cobertura del 28% de los recién nacidos españoles. En otras comunidades, como Murcia y País Vasco, aunque se ofertó anteriormente, dejó de realizarse en los años 1997 y 1995, respectivamente. Los datos obtenidos de este cribado neonatal definen una incidencia de 1/16.100 niños10.
El hecho de que los resultados del cribado se informen en el mismo tiempo cronológico que el inicio clínico de la enfermedad, la valoración en la sala de maternidad de las formas virilizantes y la escasa incidencia total de estas formas graves han llevado a que algunos sistemas de salud con una correcta cobertura perinatal de la población, como el sistema vasco, retiren de su programa este cribado neonatal. Recientemente en este aspecto, algunos autores, como Grosse et al11, han definido una mortalidad inferior al 4% de las formas graves, y defienden un avance en la detección y la sospecha clínica. Este artículo ha suscitado una importante discusión12 y algunos autores han resaltado el interés de realizar este tipo de cribado, tal y como queda recogido en los documentos de consenso de las sociedades americana y europea de endocrinología pediátrica13,14. De cualquier manera, los casos diagnosticados, tanto de forma clínica como por cribado neonatal, requieren del diagnóstico molecular que permitirá la confirmación diagnóstica, la detección de portadores y el diagnóstico prenatal requerido en el tratamiento prenatal que es posible plantear en esta enfermedad.
Presentamos el caso de una niña afectada de una forma virilizante de HSC detectada en la etapa perinatal que presentó una heterocigosis compuesta (R356W/I172N) de mutaciones graves. Únicamente el padre resultó portador (R356W) y no se evidenció mutación alguna en la madre biológica.
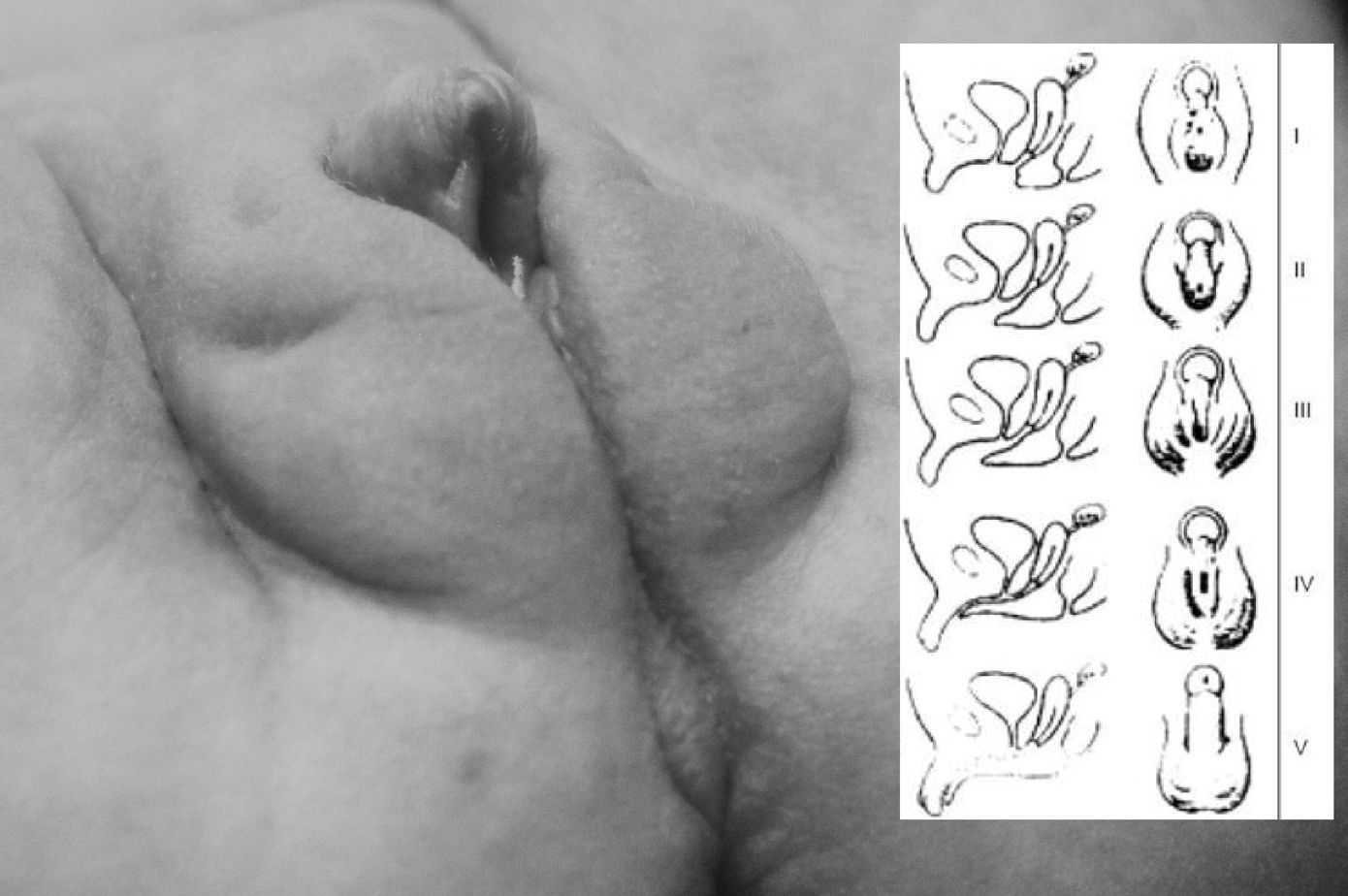
Caso clínicoMujer de origen caucasiano procedente de una primera gestación sin incidencias de 39 semanas, padres no consanguíneos. La exploración física en maternidad pasó inadvertida. Ingresó a los 20 días de vida por cuadro compatible con sepsis clínica que cursó con fiebre de 38,9°C, rechazo de tomas, cianosis y decaimiento. La analítica al ingreso mostró acidosis metabólica (pH: 7,16, presión parcial de CO2 [PCO2]: 60,8 y CO3H: 22,1), hiponatremia (Na: 123mEq/l), hiperpotasemia (7,2mEq/l) y calcio (12,5mg/dl). proteína C reactiva (PCR): 85mg/dl. En la exploración física se observó clitoromegalia, con fusión parcial de los labios mayores y el seno urogenital sin gónadas palpables (grado II–III de Prader (fig. 1)). El estudio hormonal reveló unos valores elevados de 17-hidroxiprogesterona (475,90ng/ml) y la actividad de renina en plasma (ARP) era de 38ng/ml/h (tabla 1). Presentó un cariotipo femenino normal (46XX) y genitales internos normales en ecografía pélvica.
Resumen de datos analíticos y su progresión temporal
| Veinte días | Seis meses | Doce meses | Dos años | |
| Sodio, mEq/l | 123 | 139 | 135 | 135 |
| 17-OH-P, ng/ml | 475 | 12,8 | 0,41 | 8,6 |
| Testosterona, ng/ml | 0,62 | 0,05 | – | 0,02 |
| ARP, ng/ml/h | 38 | >24 | 7,7 | 6,2 |
| Androstendiona, ng/ml | 3 | 0,04 | 0,1 | 0,16 |
| ACTH, pg/ml | 166 | – | 46 | – |
| Dosis hidrocortisona, mg/m2 | 20 | 15,8 | 15 | 16 |
| Dosis 9-alfa-flurocortisona, mg/día | 0,05 | 0,05 | 0,10 | 0,05 |
ACTH: corticotropina; ARP: actividad de renina en plasma; OH-P: hidroxiprogesterona.
Ante la sospecha de HSC se inició tratamiento con hidrocortisona (20mg/m2/día en 3 dosis), 9-alfa-fludrocortisona (0,05mg/día) y NaCl con buena evolución clínica. Se obtuvo el consentimiento informado y se procedió al estudio molecular del gen CYP21A2, que puso de manifiesto la presencia de mutaciones graves del gen (I172N y R356W) en heterocigosis compuesta (ver estudio genético más adelante).
La sepsis clínica no pudo confirmarse mediante los cultivos (sangre y líquido cefalorraquídeo [LCR]) y la paciente precisó tratamiento mineralcorticoide para mantener niveles de ARP normales para su edad (tabla 1). Durante su seguimiento no presentó episodios de descompensación con hipoglucemia ni crisis pierde sal. La dosis media requerida de hidrocortisona fue de 16mg/m2/día y 0,05–0,10mg/día de 9-alfa-fludrocortisona (tabla 1). Asimismo, se administró NaCl (1–2g/día) durante los 2 primeros años de vida.
La paciente ha mantenido una talla en percentiles 3-10 con discreto retraso en la edad ósea de 6 meses a la edad cronológica de 2 años (talla diana: 153cm). Al año de edad se realizó genitoplastia completa con buenos resultados cosméticos. En el transcurso de la elaboración de este manuscrito la madre presentó una nueva gestación de la misma pareja biológica, cuyo estudio molecular también se realizó.
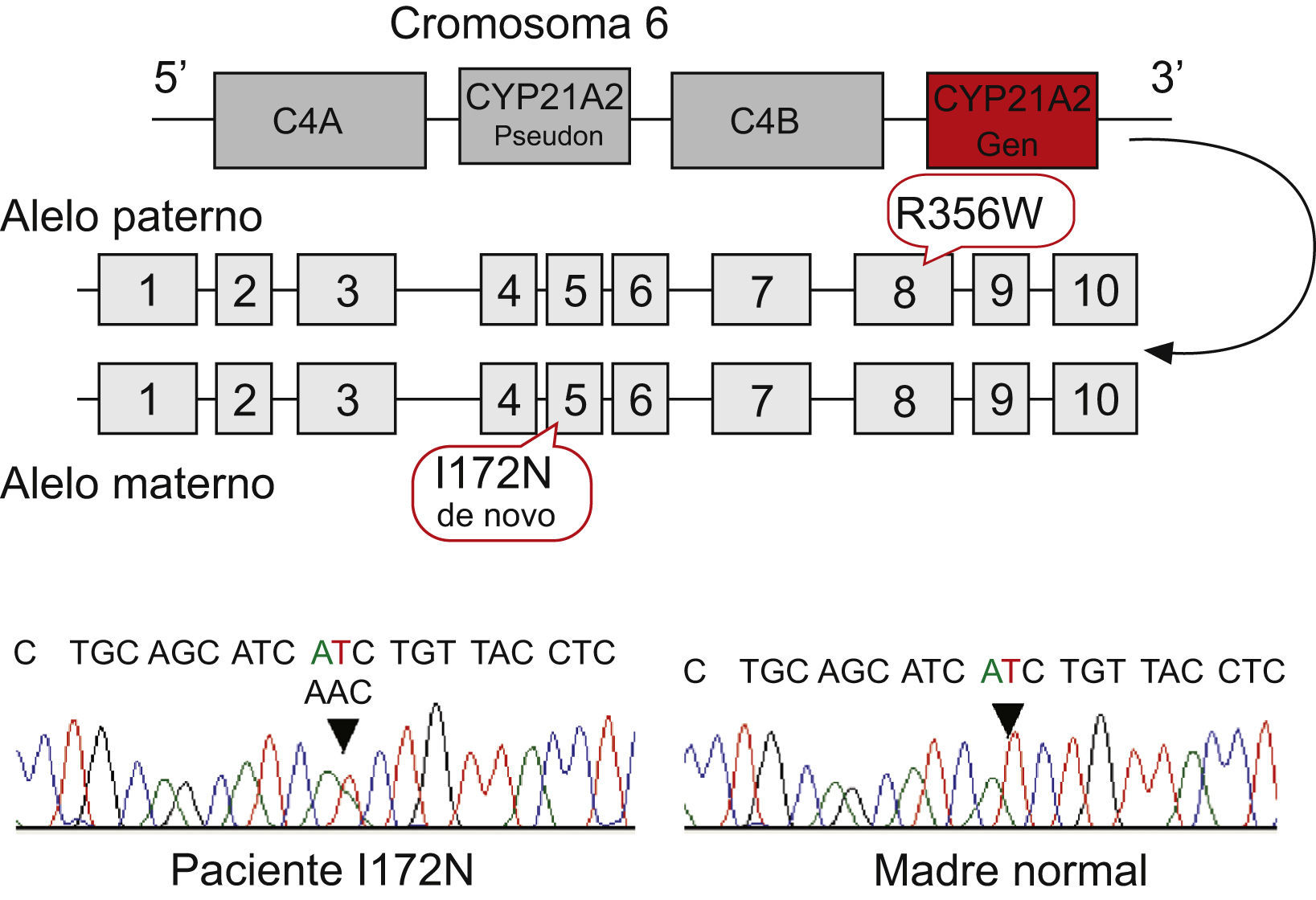
Estudio genéticoSe extrajo ADN a partir de leucocitos de sangre periférica de la paciente y de ambos padres y se procedió a la amplificación específica del gen CYP21A2, según la técnica recogida en Ezquieta et al6. El análisis directo del gen se realizó mediante estudio de deleciones y conversiones por técnica de Southern, y el cribado de las mutaciones puntuales recurrentes (P30L, 655G, deleción 8 pb, I172N, triple mutación del exón 6, V281L, 306insT, Q318X, R356W y P453S) se realizó mediante reacción en cadena de la polimerasa e hibridación específica del alelo. El estudio básico incluye también la mutación R426H, que se ha incorporado en nuestro cribado (Ezquieta et al, manuscrito en preparación) porque permite completar la caracterización de los alelos de las formas virilizantes simples. Para la secuenciación directa se utilizaron dideoxinucleótidos fluorescentes y un ABIPrism 3100 de la línea instrumental de secuenciación del Hospital Gregorio Marañón, como se describe en Ezquieta et al7. El estudio se completó con el análisis indirecto de microsatélites del cromosoma 6, asociados al gen CYP21A2, D6S273 y D6S439 y el microsatélite intragénico del factor de necrosis tumoral (TNF)7. También se analizaron marcadores de los cromosomas X e Y de la región seudoautosómica (DXY233 y DXY234) y microsatélite intragénico del gen SHOX mediante la utilización de los cebadores recogidos en Ezquieta et al25.
El árbol familiar se muestra en la figura 2 que detalla la segregación de alelos en la familia tanto en el análisis directo (mutaciones detectadas) como en el análisis indirecto (microsatélites asociados). El cribado básico de mutaciones mostró que el caso índice presentaba 2 mutaciones graves: I172N y R356W. El estudio de segregación alélica demostró el origen paterno de la mutación R356W y la ausencia de la mutación I172N en el ADN materno. La secuenciación directa de los alelos en una nueva muestra confirmó la presencia de la mutación en el caso índice y su ausencia en la madre (fig. 2). El genotipo detectado en la paciente se corresponde con el cuadro clínico virilizante de la enfermedad, ya que la mutación I172N asocia una actividad enzimática residual que evita la pérdida salina, aunque sí puede existir descompensación electrolítica y respuesta de renina.
El estudio complementario de marcadores tipo microsatélite de la región de antígenos de histocompatibilidad (HLA) D6S273, D6S439 y polimorfismo intrónico del gen TNF pudo evidenciar la procedencia de los alelos paterno y materno (fig. 3). El estudio confirmatorio de segregación se completó con los marcadores DXY233, DXY234 y el marcador intragénico del gen SHOX25 de la región seudoautosómica de los cromosomas sexuales.
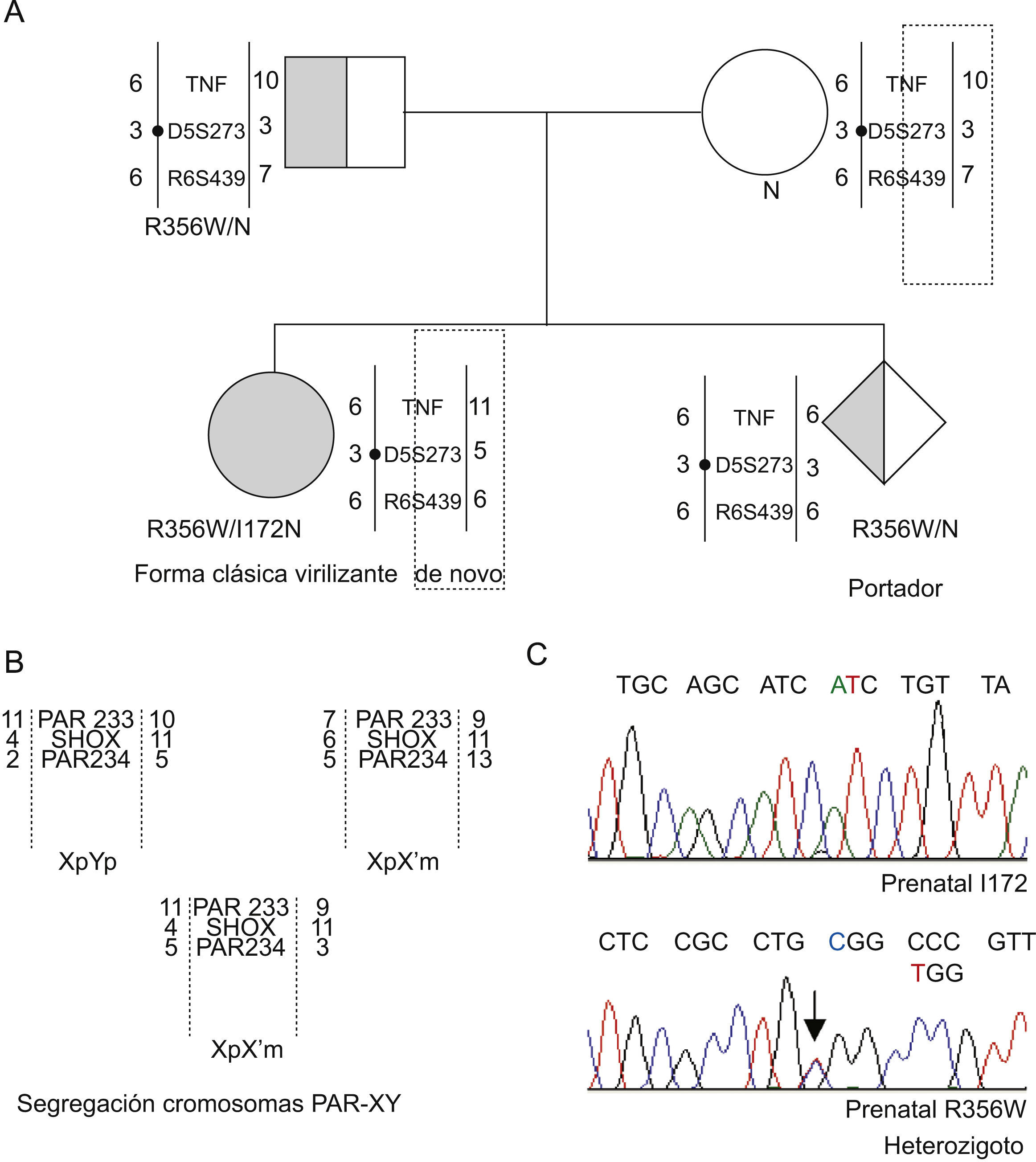
A) Representación del árbol familiar con la segregación de los alelos CYP21A2, tanto directa como indirecta. Los cromosomas se esquematizan como barras en las que se indican de forma numérica los distintos tipos de alelos de los microsatélites D6S273, D6S439 y el marcador intrónico de TNF y el haplotipo que configuran para cada uno de los cromosomas 6p del grupo familiar. B) También se recoge la segregación informativa de los cromosomas sexuales mediante la utilización de los marcadores de la región PAR, DXY233, DXY234 y microsatélite intragénico del gen SHOX. Estudios de secuenciación parcial de los exones 4 y 8 de la muestra prenatal que documentan que el nuevo hijo es únicamente portador de la mutación 356W.
Posteriormente a este estudio inicial, y conocida una nueva gestación de la madre, se decidió realizar el estudio genético prenatal mediante amniocentesis y cultivo de amniocitos para obtención de ADN. Al utilizar las técnicas moleculares descritas, se documentó que este feto (46XY) era portador de una de las 2 mutaciones graves del caso índice: la R356W (origen paterno). El alelo materno heredado por el feto es el alelo normal (fig. 3). Estos hallazgos se han confirmado tras el nacimiento y en la actualidad es un varón aparentemente sano para el 21-OHD.
DiscusiónLa detección temprana de formas virilizantes neonatales1 por el 21-OHD es importante, ya que el tratamiento adecuado da lugar a una evolución clínica más favorable. La base molecular de estas formas clínicas, también recesivas como las formas pierde sal, está constituida por 2 alelos de nula o muy baja funcionalidad denominados graves, aunque generalmente uno de ellos mantiene una mínima actividad residual, generalmente la mutación puntual I172N15–17.
Aunque el fenotipo más frecuentemente relacionado con la mutación I172N18 es el virilizante simple, se han publicado diferentes formas clínicas asociadas22,23, que incluyen formas pierde sal, virilizantes simples y formas no clásicas; la frecuencia de pérdida salina es del 25–65%27. Esta variabilidad se debe, en la gran mayoría de los casos, a las mutaciones presentes en el segundo alelo, y el cuadro clínico es de tipo no clásico cuando la segunda mutación es de tipo leve28,29. La forma pierde sal es más frecuente en aquellos individuos que presentan una segunda mutación más grave30. La forma de presentación clínica del paciente que aquí presentamos fue de una pérdida salina, en un contexto clínico de un cuadro febril, aunque la sepsis no pudo confirmarse mediante los cultivos. La virilización, que pasó inicialmente desapercibida, fue moderada, y la ausencia de episodios de descompensación o pérdida salina durante la evolución clínica posterior serían compatibles con un déficit enzimático no completo concordante con el esperado del genotipo detectado: una mutación nula (R456W) y una alteración que deja una actividad residual (I172N). El requerimiento mineralcorticoide para el mantenimiento de los niveles de la ARP sería una característica compartida con las formas clásicas sin pérdida salina3,13.
En la actualidad, la detección de un nuevo caso clínico de la forma grave de la deficiencia puede tener su clave en el cribado neonatal13,14 o mediante una detallada exploración en salas de maternidad/neonatos. La sospecha se confirma mediante el diagnóstico apoyado por los estudios genéticos y se debe analizar también a los familiares de primer grado11,12. Ello no sólo facilita una mejor caracterización del genotipo, ya que permite segregar las mutaciones detectadas, sino que establece la situación de portadores en los miembros de la familia y posibilita un futuro consejo genético. Debe tenerse en especial consideración que es posible evitar la virilización de un nuevo feto femenino afectado mediante tratamiento prenatal1,2.
Se requiere un estudio completo, como el presentado, para poder documentar el posible origen de novo de la mutación detectada. Tanto los marcadores microsatélite del complejo HLA en el autosoma 6 como los marcadores no autosómicos de la región PAR de los cromosomas sexuales X e Y documentaron una adecuada segregación de alelos en esta familia. El estudio de segregación de los alelos parentales en las enfermedades recesivas ha llevado a documentar mutaciones en sólo uno de ellos, y no es infrecuente en la literatura médica la identificación del gen afectado en la madre, pero no así en el padre. La consiguiente sospecha de no paternidad biológica suele resolverse mediante estudios de segregación de marcadores polimórficos informativos. Los estudios en bancos de sangre24 han mostrado que la duda de la posible paternidad biológica debe tenerse en consideración para documentar mutaciones de novo.
Otra causa de aparentes alelos de novo detectados en la genotipificación de casos afectados de HSC y que en la actualidad no resulta infrecuente es el antecedente de reproducción asistida con donación de esperma u óvulos31. La elevada frecuencia de portadores en población general y la posible asociación de la infertilidad en este cuadro clínico han podido contribuir a este hecho.
El posterior embarazo de la madre del caso índice nos ha permitido estudiar genéticamente al feto, aunque dentro del protocolo de valoración de necesidad de tratamiento prenatal con dexametasona15 éste se consideró inadecuado, ya que no se cumplía el supuesto de riesgo 1:4 de feto afectado. El estudio molecular se realizó en cultivo de amniocitos y mostró que el feto era sólo portador de una de las 2 mutaciones graves del caso índice (la de origen paterno [R356W]). El alelo materno (no portador de mutación) presente en la muestra fetal resultó ser el no heredado por el caso índice, por lo que no pudo quedar documentado si otras células germinales tampoco presentaban la mutación descrita.
Aunque las mutaciones de novo del gen CYP21A2 se han detectado en estudios con amplias cohortes, se ha estimado que son muy infrecuentes (<1:5.000) en la población general. Menos del 1% de las mutaciones CYP21 inactivadoras tienen este origen19–21, por lo que su descripción en la literatura médica es rara. Tajima et al26 y Collier et al28 describieron las primeras mutaciones de novo en 1993, precisamente en 2 pacientes con mutación del gen CYP21A2. Baumgarner-Parzer et al27 describieron uno de los casos más recientemente publicados. Todas las mutaciones de novo descritas hasta el momento, incluida la que aquí se documenta, se producen por los mecanismos que dan lugar a las distintas variantes alélicas (recombinación asimétrica y conversión génica) en el proceso de gametogénesis (meiosis) y su posterior fusión en la fecundación, por lo que no se puede descartar un mosaicismo germinal en estos casos.
En el caso de novo publicado por Collier et al28 también se evidenció el origen materno de la mutación mediante el estudio de secuenciación de los genes y la comparación de los diferentes nucleótidos, pero no se realizó un estudio de microsatélites como el presentado. Tajima et al26 presentaron un caso de mutación de novo en una paciente afectada de forma clásica detectada en el cribado neonatal con estudio negativo de varios hermanos, pero tampoco se realizó estudio de microsatélites para valoración de la segregación parental. En el caso más reciente de Baumgarner-Parzer et al27 se presentó a una paciente que mostraba una heterocigosis compuesta de I172N de novo, como el caso presentado, y la mutación de procesamiento del ARN mensajero 655G del intron 2. Al igual que nuestro caso, no desarrolló pierde sal clínicamente evidente, aunque la ambigüedad genital fue mayor.
En resumen, el caso que presentamos ilustra la importancia de realizar el estudio genético no sólo del paciente, sino también de los padres, anteriormente llamados “portadores obligados”. Este hecho podrá tener importantes implicaciones, no sólo medicolegales, sino también de planificación familiar.
Al proyecto FIS PI061179 de uno de los autores (B.E.) de este artículo, sin el cual este trabajo no hubiera sido posible. Este caso ha sido presentado32 en su vertiente clínica por estos autores.
