



El síndrome de Noonan (SN) es una enfermedad de origen genético relativamente frecuente cuyas manifestaciones fundamentales son la talla baja, la cardiopatía congénita y un fenotipo facial característico.
La causa del síndrome de Noonan y de otras enfermedades clínicamente solapadas como el síndrome de Noonan con lentiginosis múltiple (anteriormente llamado síndrome LEOPARD), el cardiofaciocutáneo o el síndrome de Costello, son mutaciones en genes que codifican para proteínas de la vía de señalización de las RAS-MAPKinasas. Debido a este sustrato común este grupo de enfermedades son denominadas colectivamente «rasopatías». A pesar de los avances genéticos de las últimas décadas, cerca de 20% de pacientes no tienen causa genética identificada, y el diagnóstico sigue siendo clínico.
El síndrome de Noonan se caracteriza por una alta heterogeneidad clínica y genética, con afectación variable, y cambiante con la edad, de múltiples órganos y sistemas. Debido a esta variabilidad es fundamental que los médicos involucrados en su cuidado estén familiarizados con sus manifestaciones y conozcan las recomendaciones de seguimiento, incluido el seguimiento del crecimiento y desarrollo. Hasta la fecha los escasos datos de crecimiento con GH a talla adulta dan resultados de ganancia de talla moderados, semejantes a los obtenidos en el síndrome de Turner.
La hiperactivación de la vía RAS-MAPK como base común de esta familia de enfermedades brinda una oportunidad única para el desarrollo de tratamientos dirigidos a la etiología de estos trastornos.
Noonan syndrome (NS) is a relatively common genetic condition characterised by short stature, congenital heart defects, and distinctive facial features.
NS and other clinically overlapping conditions such as NS with multiple lentigines (formerly called LEOPARD syndrome), cardiofaciocutaneous syndrome, or Costello syndrome, are caused by mutations in genes encoding proteins of the RAS-MAPKinases pathway. Because of this shared mechanism, these conditions have been collectively termed «RASopathies». Despite the recent advances in molecular genetics, nearly 20% of patients still lack a genetic cause, and diagnosis is still made mainly on clinical grounds.
NS is a clinically and genetically heterogeneous condition, with variable expressivity and a changing phenotype with age, and affects multiple organs and systems. Therefore, it is essential that physicians involved in the care of these patients are familiarised with their manifestations and the management recommendations, including management of growth and development. Data on growth hormone treatment efficacy are sparse, and show a modest response in height gains, similar to that observed in Turner syndrome.
The role of RAS/MAPK hyper-activation in the pathophysiology of this group of disorders offers a unique opportunity for the development of targeted approaches.
El síndrome de Noonan (SN) es un trastorno genético de herencia autosómica dominante caracterizado por una triada fenotípica característica: anomalías craneofaciales que confieren un fenotipo facial típico, cardiopatía congénita y talla baja1. Descrito por la doctora Jacqueline Noonan hace más de 50 años, es un trastorno relativamente frecuente, con una incidencia que se estima entre uno por cada 1.000 a 2.500 recién nacidos vivos. Desde el año 2001 se conoce que las mutaciones en genes que codifican para proteínas implicadas en la vía de señalización intracelular de las RAS-MAPK (Mitogen Activated Protein Kinases) causan el SN y otros trastornos genéticos con fenotipos similares como el síndrome de Costello, el síndrome cardiofaciocutáneo, el síndrome de Noonan con lentiginosis múltiple (SNLM, anteriormente conocido como síndrome LEOPARD) o la neurofibromatosis tipo 1. El solapamiento clínico de estas entidades y el sustrato molecular que comparten ha propiciado que sean agrupadas bajo la denominación de «síndromes neuro-cardio-facio-cutáneos» o más genéricamente, «rasopatías».
El presente trabajo pretende sintetizar el conocimiento actual de esta entidad, y proponer unas pautas de actuación a seguir en estos pacientes desde la infancia a la etapa adulta.
Descripción clínicaEl SN se caracteriza por una afectación multisistémica con alta heterogeneidad y expresión clínica variable.
Rasgos craneofacialesSe caracteriza por una facies peculiar que cambia con la edad, siendo más llamativa en la niñez y más sutil en la etapa adulta2. Los rasgos característicos son facies en forma de triángulo invertido (frente ancha que se estrecha hacia la barbilla), baja implantación de las orejas con rotación posterior y hélix engrosado (90%), fisuras palpebrales descendentes (95%), hipertelorismo, ptosis palpebral, cejas arqueadas en forma de diamante, iris usualmente de color azul o verde-azulado, nariz con raíz deprimida y punta bulbosa, filtrum largo y profundo, picos altos en el labio superior (recuerda el arco de Cupido), cuello corto, y baja implantación del cabello en forma de W (55%) (fig. 1). En recién nacidos se puede observar exceso de piel nucal.
Manifestaciones cardiovasculares
La afectación cardiaca es una de las principales características. Su frecuencia se estima entre 70-80%1,3, aunque su diagnóstico prenatal es infrecuente. Se describe un amplio espectro de alteraciones siendo la más común la estenosis pulmonar frecuentemente con válvulas displásicas (50-60%), la miocardiopatía hipertrófica (20%)4, usualmente presente en etapas tempranas de la vida5, defectos del septum atrial (6-10%) y ventricular, estenosis de la rama de la arteria pulmonar, anomalías de la válvula mitral, arterias coronarias, coartación de la aorta y, menos frecuente, la tetralogía de Fallot o ductus arterioso permeable.
Se han documentado alteraciones electrocardiográficas (50%)6. A menudo muestran complejos QRS anchos con un patrón predominantemente negativo en las derivaciones precordiales izquierdas y la desviación del eje izquierdo con ondas Q gigantes, incluso, con estructuras cardiacas normales. Las arritmias son infrecuentes7.
Manifestaciones en crecimiento y desarrolloEl crecimiento prenatal no suele verse afectado, aunque hay una mayor prevalencia de recién nacido pequeño para la edad gestacional que en la población general8.
Los niños con SN desarrollan un patrón de hipocrecimiento postnatal caracterizado por un crecimiento prepuberal en percentil 3, retraso de inicio puberal con escaso estirón y una talla adulta en torno a -2 desviaciones estándar. El retraso puberal es común con una edad media al inicio de la pubertad de 13,4 años (10,8-16,4 años) en los niños y 13 años (10,9-15 años) en las niñas. La edad ósea tiene un retraso medio de 2 años. Se han desarrollado curvas de crecimiento específicas para niños con SN9, ninguna de ellas en población española.
Algunos autores han encontrado perfiles hormonales sugestivos de insensibilidad a la hormona de crecimiento en pacientes con SN, con niveles de IGF1 e IGFBP-3 disminuidos y respuestas de hormona de crecimiento al estímulo en el límite alto10. Otros estudios sugieren que la hiperactivación de RAS-MAPK podría afectar a la diferenciación de los condrocitos durante el crecimiento óseo, mediante un mecanismo independiente de IGF111.
Manifestaciones nutricionales y alteraciones en la homeostasis energéticaEn la etapa neonatal presentan dificultades para la alimentación mostrando succión débil, tiempo de alimentación prolongado, episodios esporádicos de náusea y vómito que, en algunos casos (25%), requieren alimentación por sonda nasogástrica, y reflujo gastroesofágico. Estos síntomas suelen mejorar o resolverse antes de los 15 meses de vida12,13.
Varios estudios han documentado que los pacientes con SN y otras rasopatías tienen tendencia al bajo peso, no solo en los primeros años de vida, sino a lo largo de la vida. Se ha descrito un modelo animal con la variante p.Thr468Met (típica del SNLM) con un fenotipo metabólico que incluye defectos de la adipogénesis, aumento del gasto energético y actividad mitocondrial, mejoría de la sensibilidad a la insulina y resistencia a dieta obesógena14. Las implicaciones de estos hallazgos son inciertas, si bien cabe esperar que investigaciones futuras aporten datos en relación con esta alteración de la homeostasis energética.
Trastornos genitourinarios y renalesLa criptorquidia se describe en 60-80% de los varones12. Varios estudios han documentado valores más altos de LH y FSH y niveles más bajos de inhibina B y hormona antimülleriana (HAM) en pacientes con SN frente a la población general, con predominio de un patrón hormonal de disfunción de células de Sertoli, tanto en aquellos con antecedente de criptorquidia como en aquellos que no la tuvieron15. Esta hipótesis se ve reforzada por el papel crítico de SHP2 en el mantenimiento de la función celular de Sertoli. Los estudios muestran, asimismo, mayor frecuencia de infertilidad en los varones con SN.
Por el contrario, se considera que las mujeres con SN tienen una fertilidad normal (lo que explicaría que en los casos familiares la transmisión más frecuente es materna).
Existen alteraciones renales en 10% de los casos (doble sistema colector, riñón único, estenosis pieloureteral y dilatación de la pelvis renal)16.
Hipotiroidismo y trastornos autoinmunesLa mayoría de estudios encuentran autoinmunidad tiroidea más frecuente que en población general13, aunque similar o discretamente más alta de hipotiroidismo subclínico17. Otras manifestaciones descritas son enfermedad celíaca, lupus eritematoso sistémico, vitíligo y uveítis anterior.
Trastornos musculoesqueléticosPresentan deformidad torácica (pectus excavatum inferior y carinatum superior) en 70-95%, así como separación de los pezones, cúbito valgo, genu valgo18, escoliosis (10-15%) y otras anomalías espinales menos frecuentes: cifosis, espina bífida, anomalías vertebrales y costales. La piel del cuello se aprecia redundante y el trapecio prominente. La hiperextensibilidad articular es común13.
Se ha descrito en niños una densidad mineral ósea de cuerpo total más baja en comparación con los controles de edad, sexo, altura y etnia. Sin embargo, la frecuencia de fracturas a esa edad no parece ser superior a la de la población general. La frecuencia de fracturas en el adulto con SN es desconocida, por lo que la trascendencia clínica de estos hallazgos es incierta, y debe ser contrastada con el desarrollo de estudios longitudinales. El análisis de histomorfometría del tejido óseo en niños con SN y modelo de ratón de Noonan podría aclarar aún más la relación entre la vía RAS-MAPK y la homeostasis esquelética19.
Trastornos oncohematológicosLa tendencia a hematomas y sangrado es frecuente, especialmente en la infancia, aunque las hemorragias graves son raras (3%). Se han descrito tiempo prolongado de sangrado, déficit de factores VIII, XI y XII, plaquetopenia y defectos de la función plaquetaria, de forma aislada o combinada20. A menudo no hay correlación entre los resultados de las pruebas de coagulación y la tendencia al sangrado.
El SN, como el resto de rasopatías, presenta un riesgo aumentado de neoplasias hematológicas y tumores sólidos que, en la actualidad, se estima 8,1 veces superior (IC 95% 3,5-16) al de la población general21. Destacan los gliomas (tumores neuroepiteliales disembrioplásticos), leucemia linfoblástica aguda, neuroblastoma y rabdomiosarcoma. Las sustituciones en el codón 61 y la sustitución 218C >T (p.Thr73Ile) en el gen PTPN11, la variante p.Thr58Ile en KRAS, y las alteraciones en el gen CBL se asocian a una mayor frecuencia de leucemia mielomonocítica juvenil (LMMJ), un trastorno mieloproliferativo grave22. Los casos de LMMJ tienden a tener una presentación precoz y un curso benigno, con casos descritos de resolución espontánea. Un estudio reciente documenta, sin embargo, una mortalidad más alta de la esperada en el SN atribuible a casos de LMMJ neonatal grave23 y sugiere que este trastorno podría pasar desapercibido dada la evolución fatal de estos casos.
Manifestaciones neurológicas, problemas cognitivos y conductualesLa capacidad cognitiva se encuentra dentro del rango de la normalidad hasta en 80% de los casos24 con un coeficiente intelectual que varía entre 70 y 1202,25. Se han documentado deficiencias en el reconocimiento de las emociones y la incapacidad para expresarlas verbalmente (alexitimia). Se describen trastornos del estado de ánimo, dificultades de comunicación e interacción social, déficit de atención y trastorno de hiperactividad13. Las deficiencias del lenguaje son más comunes y cuando están presentes, están asociadas con un alto riesgo de dificultades de lectura y ortografía24.
Manifestaciones orodentalesSe describe paladar alto y arqueado (55-100%), maloclusión dental (50-67%), problemas en la articulación temporo-mandibular (72%) y micrognatia. Se ha descrito maloclusión de clase II y una mayor incidencia de mordida abierta y cruzada posterior en comparación con la población general. Además, se observan dientes permanentes sin erupción y dientes deciduos sumergidos y supernumerarios26. El tumor mandibular de células gigantes es una manifestación infrecuente, pero altamente sugestiva del síndrome.
Trastornos linfáticosEl linfedema está presente en menos de 20%, sin embargo con morbilidad significativa. El linfedema periférico puede ocurrir desde el nacimiento (dorso de manos y pies) pudiendo resolverse en los primeros años de vida27. Otros trastornos menos frecuentes son: hidropesía fetal, linfangiectasia pulmonar, intestinal o testicular, quilotórax y ascitis quilosa, vasos linfáticos hipoplásicos inguinales e ilíacos, aplasia o ausencia del conducto torácico, linfedema en escroto y vulva16,28.
Alteraciones cutáneasPresentan nevus pigmentados (25%), manchas café con leche (10%) y léntigos. La queratosis pilaris es común (parte superior de los brazos), cuando se presenta en la cara conlleva a ausencia de cejas29. La piel es hiperelástica, el cabello grueso y rizado y las uñas distróficas. En los dedos se observan almohadillas fetales (67%)13.
Problemas de vista y audiciónPresentan alteración oftalmológica (95%): estrabismo (40-65%), defecto de refracción (60%), ambliopía (33%) o nistagmus (10%). Desarrollan cambios en el segmento anterior (60%), entre ellas, la catarata. Presentan drusas ópticas (20%), hipoplasia del nervio óptico y coloboma13,30. La pérdida de audición debido a otitis media es una complicación frecuente (15-40%). La pérdida auditiva neurosensorial es menos común31. Ocasionalmente se han descrito anomalías vestibulares y estructurales del oído interno.
Pronóstico y calidad de vidaLos datos a largo plazo son escasos. Se ha descrito que las dificultades de alimentación en la infancia fueron un marcador temprano de retraso en el lenguaje. En un estudio un tercio de los adultos habían asistido a una escuela para niños con dificultades para el aprendizaje y 20% a escuela convencional con refuerzo educativo. Obtuvieron un grado de certificación (secundaria o nivel educativo más alto) en 43% de los casos. En cuanto a la valoración de la calidad de vida relacionada con la salud, los resultados publicados no difieren de la población general32.
Los adultos requieren seguimiento cardiológico a largo plazo. La ausencia de síntomas, gasto cardíaco y presión arterial pulmonar normal, así como presión ventricular derecha menor de 100 mm Hg se asocian a pronóstico favorable33. Aquellos con miocardiopatía hipertrófica tienen la misma tasa de mortalidad anual que aquellos que no presentan SN. La tasa de mortalidad fue de 9% con una edad media de 60 años15.
Genética del síndrome de Noonan y las rasopatíasLa vía RAS-MAPKLa vía de las RAS-MAPKinasas es una vía de señalización intracelular ampliamente conocida que vehiculiza la señal de ligandos extracelulares como hormonas, citokinas y factores de crecimiento hasta producir la transcripción en el núcleo celular, participando en procesos de proliferación, diferenciación celular y apoptosis. RAS funciona como un interruptor molecular que estimula la activación secuencial de la cascada de las MAPK, tres niveles de kinasas (RAF, MEK y ERK) que tras una sucesión de fosforilaciones acaban produciendo su efecto mediante modificación transcripcional (fig. 2). La mayoría de las mutaciones descritas en estos genes son de «ganancia de función» y, en la actualidad, se considera que las rasopatías son consecuencia de un aumento de función, o al menos de una desregulación de la vía RAS-MAPK. La herencia es autosómica dominante, aunque recientemente se han descrito casos debidos a variantes bialélicas en LZTR1 que se trasmitirían mediante herencia autosómica recesiva.
 que favorecen el cambio conformacional de la proteína RAS inactiva unida a GDP, a la forma activa unida a GTP. RAS-GTP activa consecuentemente las distintas isoformas de RAF (RAF1, BRAF), MEK (MEK1, MEK2) y, por último, ERK.")
Cascada de señalización RAS-MAPK. Tras la unión del ligando a los receptores celulares, el fragmento intracelular de estos se fosforila reclutando proteínas adaptadoras como GRB2, formando un complejo con factores de intercambio de guaninas (como SOS) que favorecen el cambio conformacional de la proteína RAS inactiva unida a GDP, a la forma activa unida a GTP. RAS-GTP activa consecuentemente las distintas isoformas de RAF (RAF1, BRAF), MEK (MEK1, MEK2) y, por último, ERK.
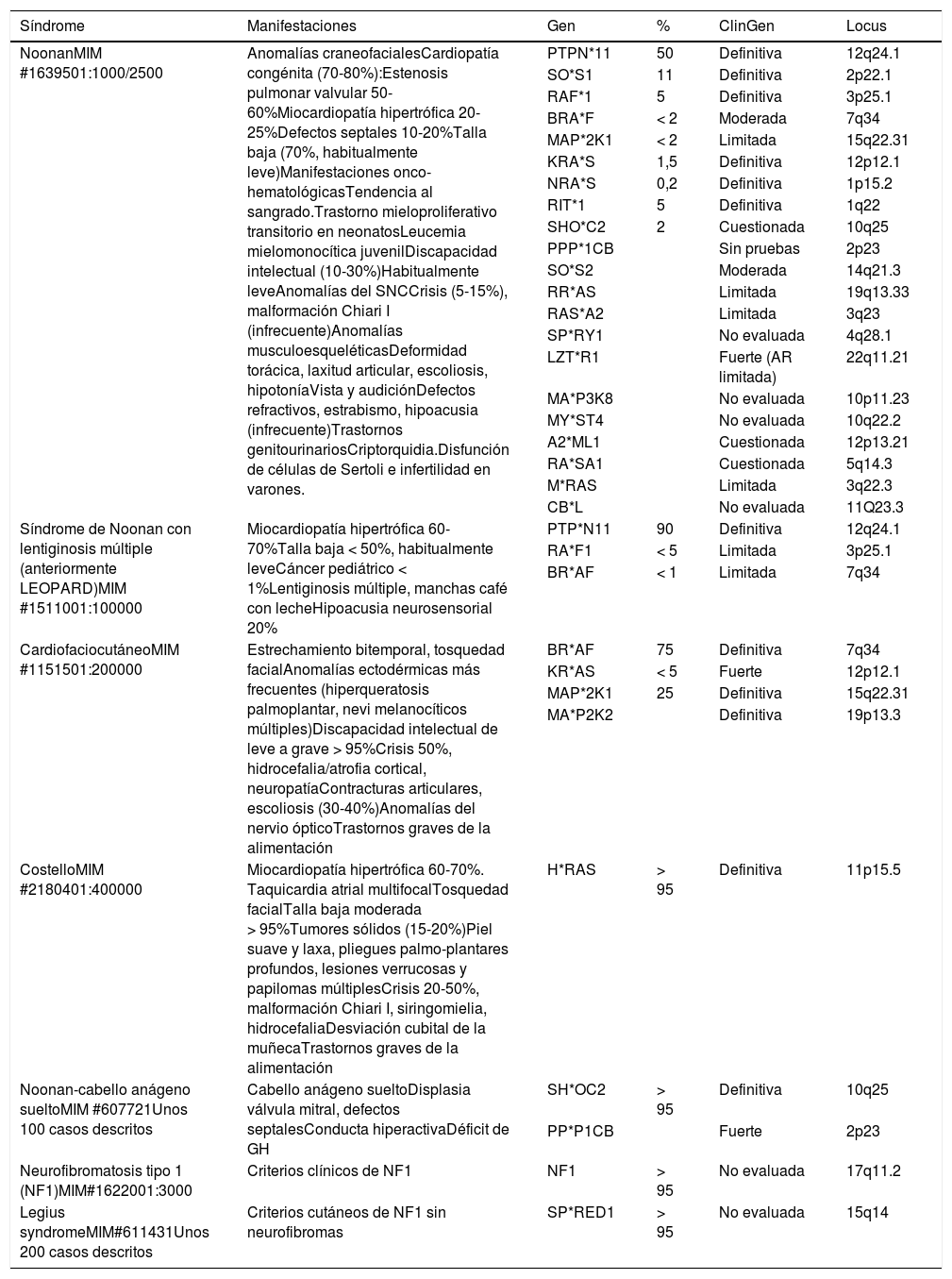
Hasta la fecha se han descrito mutaciones en más de 20 genes distintos en el SN, la rasopatía con mayor heterogeneidad de locus. Sin embargo se trata de un trastorno monogénico, y hasta la fecha la presencia de mutaciones en más de un gen en pacientes con rasopatías sigue siendo una rareza. La tabla 1 resume los genes implicados conocidos en el momento actual. Dado el número creciente de genes implicados y la multiplicidad de laboratorios que ofrecen estudios mediante secuenciación masiva, se hace necesario un esfuerzo para confirmar la implicación de los genes identificados, así como de las variantes identificadas en cada gen. El panel de expertos en rasopatías de la Clinical Genome Resource (ClinGen), fundada por National Institutes of Health, ha publicado recientemente una revisión de 19 genes implicados en rasopatías que va en esa dirección34 y aporta una clasificación de la fuerza de la evidencia clínica y experimental entre los distintos genes y los fenotipos a los que se han asociado.
Relación de síndromes relacionados con alteraciones en genes de la vía RAS-MAPK
| Síndrome | Manifestaciones | Gen | % | ClinGen | Locus |
|---|---|---|---|---|---|
| NoonanMIM #1639501:1000/2500 | Anomalías craneofacialesCardiopatía congénita (70-80%):Estenosis pulmonar valvular 50-60%Miocardiopatía hipertrófica 20-25%Defectos septales 10-20%Talla baja (70%, habitualmente leve)Manifestaciones onco-hematológicasTendencia al sangrado.Trastorno mieloproliferativo transitorio en neonatosLeucemia mielomonocítica juvenilDiscapacidad intelectual (10-30%)Habitualmente leveAnomalías del SNCCrisis (5-15%), malformación Chiari I (infrecuente)Anomalías musculoesqueléticasDeformidad torácica, laxitud articular, escoliosis, hipotoníaVista y audiciónDefectos refractivos, estrabismo, hipoacusia (infrecuente)Trastornos genitourinariosCriptorquidia.Disfunción de células de Sertoli e infertilidad en varones. | PTPN*11 | 50 | Definitiva | 12q24.1 |
| SO*S1 | 11 | Definitiva | 2p22.1 | ||
| RAF*1 | 5 | Definitiva | 3p25.1 | ||
| BRA*F | < 2 | Moderada | 7q34 | ||
| MAP*2K1 | < 2 | Limitada | 15q22.31 | ||
| KRA*S | 1,5 | Definitiva | 12p12.1 | ||
| NRA*S | 0,2 | Definitiva | 1p15.2 | ||
| RIT*1 | 5 | Definitiva | 1q22 | ||
| SHO*C2 | 2 | Cuestionada | 10q25 | ||
| PPP*1CB | Sin pruebas | 2p23 | |||
| SO*S2 | Moderada | 14q21.3 | |||
| RR*AS | Limitada | 19q13.33 | |||
| RAS*A2 | Limitada | 3q23 | |||
| SP*RY1 | No evaluada | 4q28.1 | |||
| LZT*R1 | Fuerte (AR limitada) | 22q11.21 | |||
| MA*P3K8 | No evaluada | 10p11.23 | |||
| MY*ST4 | No evaluada | 10q22.2 | |||
| A2*ML1 | Cuestionada | 12p13.21 | |||
| RA*SA1 | Cuestionada | 5q14.3 | |||
| M*RAS | Limitada | 3q22.3 | |||
| CB*L | No evaluada | 11Q23.3 | |||
| Síndrome de Noonan con lentiginosis múltiple (anteriormente LEOPARD)MIM #1511001:100000 | Miocardiopatía hipertrófica 60-70%Talla baja < 50%, habitualmente leveCáncer pediátrico < 1%Lentiginosis múltiple, manchas café con lecheHipoacusia neurosensorial 20% | PTP*N11 | 90 | Definitiva | 12q24.1 |
| RA*F1 | < 5 | Limitada | 3p25.1 | ||
| BR*AF | < 1 | Limitada | 7q34 | ||
| CardiofaciocutáneoMIM #1151501:200000 | Estrechamiento bitemporal, tosquedad facialAnomalías ectodérmicas más frecuentes (hiperqueratosis palmoplantar, nevi melanocíticos múltiples)Discapacidad intelectual de leve a grave > 95%Crisis 50%, hidrocefalia/atrofia cortical, neuropatíaContracturas articulares, escoliosis (30-40%)Anomalías del nervio ópticoTrastornos graves de la alimentación | BR*AF | 75 | Definitiva | 7q34 |
| KR*AS | < 5 | Fuerte | 12p12.1 | ||
| MAP*2K1 | 25 | Definitiva | 15q22.31 | ||
| MA*P2K2 | Definitiva | 19p13.3 | |||
| CostelloMIM #2180401:400000 | Miocardiopatía hipertrófica 60-70%. Taquicardia atrial multifocalTosquedad facialTalla baja moderada > 95%Tumores sólidos (15-20%)Piel suave y laxa, pliegues palmo-plantares profundos, lesiones verrucosas y papilomas múltiplesCrisis 20-50%, malformación Chiari I, siringomielia, hidrocefaliaDesviación cubital de la muñecaTrastornos graves de la alimentación | H*RAS | > 95 | Definitiva | 11p15.5 |
| Noonan-cabello anágeno sueltoMIM #607721Unos 100 casos descritos | Cabello anágeno sueltoDisplasia válvula mitral, defectos septalesConducta hiperactivaDéficit de GH | SH*OC2 | > 95 | Definitiva | 10q25 |
| PP*P1CB | Fuerte | 2p23 | |||
| Neurofibromatosis tipo 1 (NF1)MIM#1622001:3000 | Criterios clínicos de NF1 | NF1 | > 95 | No evaluada | 17q11.2 |
| Legius syndromeMIM#611431Unos 200 casos descritos | Criterios cutáneos de NF1 sin neurofibromas | SP*RED1 | > 95 | No evaluada | 15q14 |
ClinGen: Evaluación del grado de asociación de los genes a las distintas rasopatías por el panel de expertos en rasopatías de la Clinical Genome Resource34 de acuerdo con la clasificación semicuantitativa de Strande de las pruebas genéticas y experimentales: Definitiva (12-18 y constatado en el tiempo); fuerte (12-18); moderada (7-11); limitada (1-6); sin pruebas (0); cuestionada (casos descritos tras la publicación original que cuestionan el papel del gen en la causa de la enfermedad, sin refutarla); refutada. No evaluada: genes no incluidos en la evaluación de la Clinical Genome Resource. AR: herencia autosómica recesiva: se han descrito casos infrecuentes de síndrome de Noonan con afectación bialélica de LZTR1 que parecen transmitirse con este patrón de herencia.
Se han encontrado múltiples correlaciones entre genotipo y fenotipo en el SN, si bien no hay manifestaciones fenotípicas exclusivas de un genotipo, probablemente como consecuencia de factores genéticos y epigenéticos que influyen en la penetrancia y la expresividad. La tabla 2 resume algunas de las asociaciones más aceptadas.
Correlación genotipo-fenotipo
| Correlación gen-fenotipo | |
|---|---|
| Gen | Manifestaciones clínicas asociadas |
| PTPN11 | Facies típica. Estenosis pulmonar valvular. Tendencia a hematomas. Criptorquidia. Casos familiares |
| SOS1 | Manifestaciones cutáneas típicas del síndrome cardiofaciocutáneo (queratosis pilar, pelo escaso y/o rizado, cejas escasas)Baja frecuencia de talla baja y discapacidad intelectual |
| RAF1 | Miocardiopatía hipertrófica (a veces neonatal). Máculas pigmentadas |
| KRAS | Deterioro cognitivo, alteraciones cutáneas propias del síndrome cardiofaciocutáneo |
| BRAF | Deterioro cognitivo, alteraciones cutáneas propias del síndrome cardiofaciocutáneo |
| CBL | Mayor frecuencia de LMMJ y tumores sólidosBaja frecuencia de talla baja, cardiopatía congénita o criptorquidia |
| RIT1 | Miocardiopatía hipertrófica (a veces neonatal). LMMJ.Baja frecuencia de talla baja, afectación cutánea y discapacidad intelectual |
| Correlación variante-fenotipo | |
|---|---|
| Variante (gen) | Manifestaciones clínicas asociadas* |
| p.Ser2Gly (SHOC2) | Síndrome de Noonan con cabello anágeno suelto: cabello que se desprende fácilmente a la tracción en fase de crecimiento o anágena, más frecuencia de defectos septales y displasia de la mitral, anomalías ectodérmicas, hiperactividad, voz nasal, déficit de hormona de crecimiento |
| p.Thr468Met (PTPN11) | Síndrome de Noonan con lentiginosis múltiple: lentiginosis, miocardiopatía hipertrófica, hipoacusia, talla conservada |
| p.Thr73Ile, sustituciones en p.Asp61 (PTPN11) | Riesgo aumentado de LMMJ |
Mientras que la correlación estrecha entre un gen y un fenotipo específico es un marco conceptual atractivo, la experiencia clínica parece sugerir que la realidad es más complicada. Por eso todavía hoy la clasificación de las rasopatías en entidades nosológicas, más que por genes implicados, sigue siendo preferida.
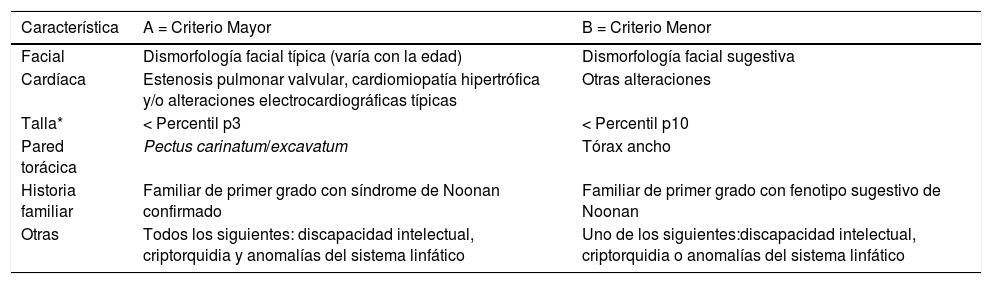
Diagnóstico y diagnóstico diferencialA pesar del desarrollo de los estudios genotípicos, el diagnóstico del SN es fundamentalmente clínico y en cerca de 20-30% de los pacientes se desconoce la causa genética. La variabilidad en la expresión clínica, el solapamiento con otras entidades, y los cambios en el fenotipo que se observan con la edad hacen que el diagnóstico sea todo un reto, particularmente en el primer año de vida. Los criterios diagnósticos uniformemente aceptados fueron desarrollados por van der Burgt (1994) y revisados en 200729 (tabla 3).
Criterios diagnósticos de síndrome de Noonan
| Característica | A = Criterio Mayor | B = Criterio Menor |
|---|---|---|
| Facial | Dismorfología facial típica (varía con la edad) | Dismorfología facial sugestiva |
| Cardíaca | Estenosis pulmonar valvular, cardiomiopatía hipertrófica y/o alteraciones electrocardiográficas típicas | Otras alteraciones |
| Talla* | < Percentil p3 | < Percentil p10 |
| Pared torácica | Pectus carinatum/excavatum | Tórax ancho |
| Historia familiar | Familiar de primer grado con síndrome de Noonan confirmado | Familiar de primer grado con fenotipo sugestivo de Noonan |
| Otras | Todos los siguientes: discapacidad intelectual, criptorquidia y anomalías del sistema linfático | Uno de los siguientes:discapacidad intelectual, criptorquidia o anomalías del sistema linfático |
En el diagnóstico diferencial se deben considerar otras rasopatías, así como otros síndromes no relacionados con la vía RAS-MAPK como el síndrome de Aarskog, el síndrome de Turner, el síndrome de Baraitser-Winter y la familia de las actinopatías. A pesar de que las otras rasopatías deben ser consideradas en el diagnóstico diferencial, todas ellas presentan una frecuencia mucho menor que el SN, con excepción de la neurofibromatosis tipo 1.
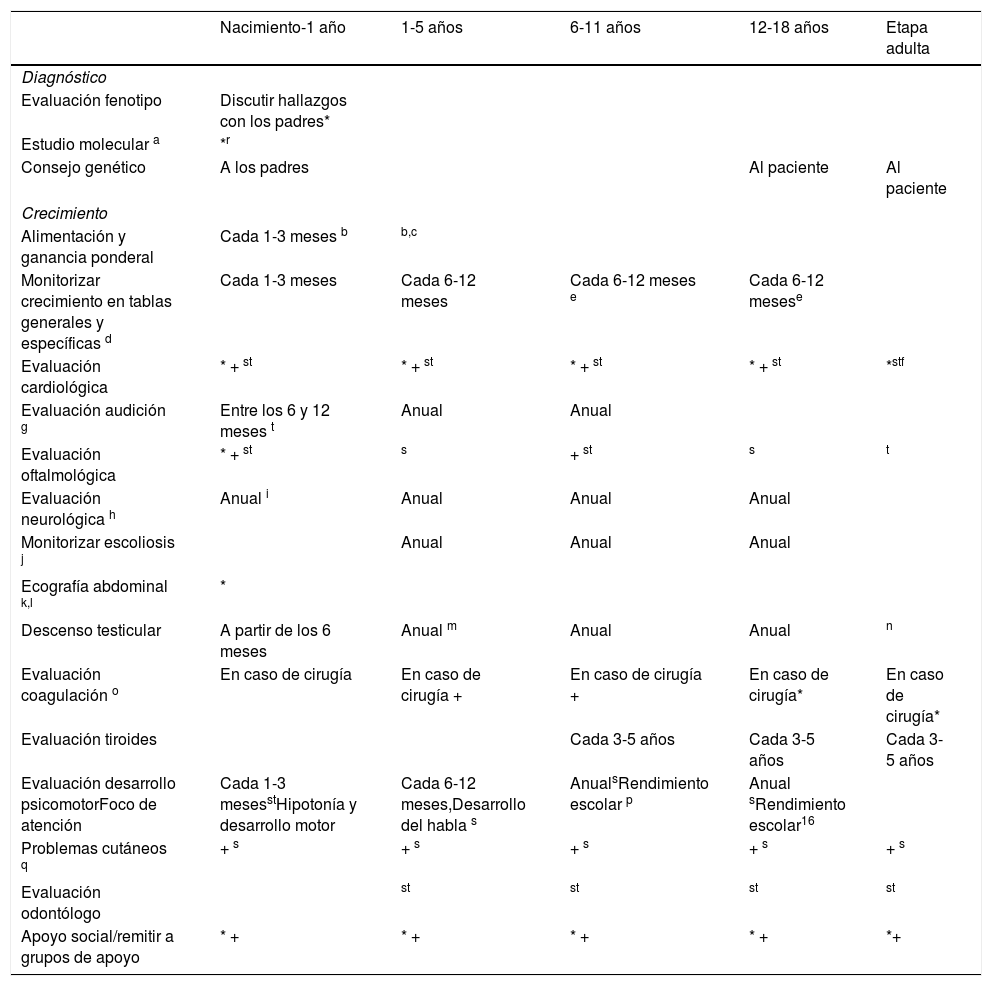
Recomendaciones de seguimientoEs importante que los diversos especialistas potencialmente implicados en la atención a estos pacientes estén familiarizados con las manifestaciones clínicas y con las posibles complicaciones asociadas al SN. Es conveniente un médico que coordine la actuación de cada uno de ellos. Esta labor puede ser desarrollada por el genetista clínico, o alternativamente por otro médico (pediatra de Atención Primaria, endocrinólogo, o cardiólogo) particularmente implicado en el seguimiento del paciente, y tiene por objeto abarcar las múltiples facetas que requieren atención y seguimiento. La tabla 49,28,29 pretende resumir las actuaciones precisas por grupos de edad.
Recomendaciones para el seguimiento
| Nacimiento-1 año | 1-5 años | 6-11 años | 12-18 años | Etapa adulta | |
|---|---|---|---|---|---|
| Diagnóstico | |||||
| Evaluación fenotipo | Discutir hallazgos con los padres* | ||||
| Estudio molecular a | *r | ||||
| Consejo genético | A los padres | Al paciente | Al paciente | ||
| Crecimiento | |||||
| Alimentación y ganancia ponderal | Cada 1-3 meses b | b,c | |||
| Monitorizar crecimiento en tablas generales y específicas d | Cada 1-3 meses | Cada 6-12 meses | Cada 6-12 meses e | Cada 6-12 mesese | |
| Evaluación cardiológica | * + st | * + st | * + st | * + st | *stf |
| Evaluación audición g | Entre los 6 y 12 meses t | Anual | Anual | ||
| Evaluación oftalmológica | * + st | s | + st | s | t |
| Evaluación neurológica h | Anual i | Anual | Anual | Anual | |
| Monitorizar escoliosis j | Anual | Anual | Anual | ||
| Ecografía abdominal k,l | * | ||||
| Descenso testicular | A partir de los 6 meses | Anual m | Anual | Anual | n |
| Evaluación coagulación o | En caso de cirugía | En caso de cirugía + | En caso de cirugía + | En caso de cirugía* | En caso de cirugía* |
| Evaluación tiroides | Cada 3-5 años | Cada 3-5 años | Cada 3-5 años | ||
| Evaluación desarrollo psicomotorFoco de atención | Cada 1-3 mesesstHipotonía y desarrollo motor | Cada 6-12 meses,Desarrollo del habla s | AnualsRendimiento escolar p | Anual sRendimiento escolar16 | |
| Problemas cutáneos q | + s | + s | + s | + s | + s |
| Evaluación odontólogo | st | st | st | st | |
| Apoyo social/remitir a grupos de apoyo | * + | * + | * + | * + | *+ |
Se recomienda que la sospecha diagnóstica y el estudio genético inicial sean orientados por un genetista clínico. La proporción esperada de resultados positivos en un paciente con diagnóstico clínico definitivo de SN es de 70-80% si se secuencian todos los genes implicados. Si se encuentra una variante patogénica es conveniente realizar el estudio en los padres para informar con precisión del riesgo de transmisión.
Remitir a consultas especializadas para evaluación de la alimentación y de la deglución si precisa. Remitir al logopeda si precisa. Los vómitos repetidos deberían estudiarse para descartar reflujo gastroesofágico o malrotación. Tratamiento con medidas antirreflujo si precisa. Los vómitos persistentes o el rechazo de la alimentación pueden precisar alimentación por sonda/gastrostomía
Usar curvas específicas del síndrome de Noonan con cautela dado que no son genotipo-específicas. Descartar déficit de hormona de crecimiento como en la población general. Valorar tratamiento con hormona de crecimiento de manera individualizada. En caso de instaurar tratamiento con hormona de crecimiento evaluación cardiológica y hematológica al inicio del mismo y monitorización estrecha.
Se debe anticipar la probabilidad de una pubertad tardía y ofrecer apoyo y asesoramiento. En caso de retraso puberal significativo (no desarrollo mamario en mujeres a los 13 años o testes menores de 4 cc con orquidómetro de Prader en varones mayores de 14 años) realizar estudio de función gonadal. Debido a la asociación con disfunción de células de Sertoli, el tamaño testicular puede no reflejar correctamente el grado de desarrollo puberal.
Evaluación con ecocardiograma aunque tenga estudio previo normal. Es recomendable una revisión cada cinco años en sujetos previamente sanos. En aquellos con cardiopatía congénita, seguimiento según indicaciones del cardiólogo. En aquellos sometidos a procedimientos quirúrgicos o valvuloplastia es esencial el seguimiento a largo plazo.
Remitir para valoración de audición en la segunda mitad del primer año. Seguimiento estrecho y manejo enérgico de las otitis para prevenir pérdidas de audición.
Alto nivel de sospecha para investigar síntomas neurológicos (considerar descartar malformación de Arnold Chiari o hidrocefalia si el paciente presenta cefalea u otros síntomas neurológicos y solicitar RNM si se sospecha). Manejo de la epilepsia como en la población general
Valorar remitir para valoración formal de desarrollo por neuropediatra, y atención temprana con terapia ocupacional y fisioterapia. Es positivo transmitir a los padres que en los pacientes con SN el retraso motor es con frecuencia consecuencia de la hipotonía, y no siempre se asocia a trastornos de aprendizaje o discapacidad intelectual en la etapa adulta.
Determinar la presencia o no de hepato-esplenomegalia. En caso de esplenomegalia realizar hemograma. En caso de hepatoesplenomegalia realizar hemograma completo y función hepática. En pacientes con variantes de alto riesgo oncogénico (sustituciones en p.61 y el cambio p.Thr73Ile en PTPN11, y la variante p.Thr58Ile en KRAS) valorar seguimiento clínico y con hemograma cada 3 meses durante el primer año de vida.
Evaluación anual para comprobar que no hay “ascenso” testicular si previamente sano. Actuación ante el maldescenso testicular como en la población general.
Se debe informar de que existe un riesgo aumentado de infertilidad en varones, incluso en aquellos sin antecedentes de criptorquidia. Remitir a una clínica de infertilidad o al endocrinólogo si precisa.
En los pacientes con síndrome de Noonan no existe buena correlación entre los resultados de las pruebas de coagulación y la tendencia al sangrado, por lo que en caso de cirugía se deberán extremar las precauciones, suspender el tratamiento con ácido acetil salicílico si lo toma antes de la intervención, y valorar contactar con hematología pediátrica para valorar el riesgo de sangrado y las medidas pertinentes para disminuir el riesgo de hemorragia.
En pacientes con estudio genético positivo es recomendable actualizar periódicamente nuestros conocimientos sobre las manifestaciones asociadas a su variante, en particular en las poco frecuentes, si es necesario con el asesoramiento de un genetista clínico experto.
Por último, en pacientes con rasopatías diferentes del SN, y paciente con SN por alteraciones en genes poco frecuentes, debe considerarse el seguimiento en unidades multidisciplinares con experiencia.
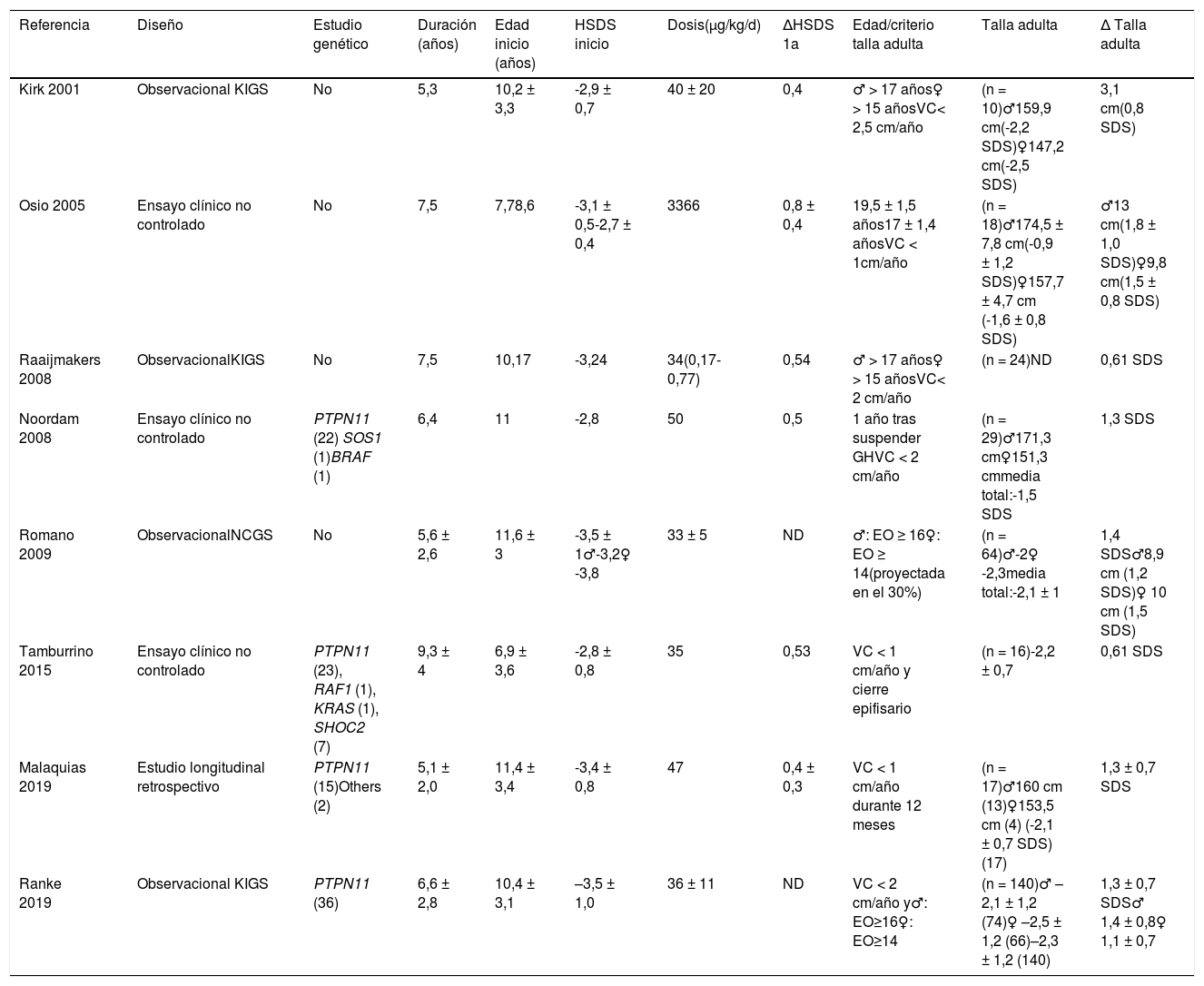
Tratamiento con hormona de crecimientoUna de las aproximaciones de tratamiento sintomático en el SN ha sido el tratamiento con hormona de crecimiento recombinante humana (rhGH). Su uso fue aprobado por la Food and Drug Administration norteamericana en 2007 y, recientemente, ha sido aprobada por la Agencia Europea del Medicamento (https://mri.cts-mrp.eu/Human/Product/Details/14639), y la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) (https://cima.aemps.es/cima/dochtml/ft/62977/FT_62977.html). Sin embargo, cabe destacar que no existen ensayos clínicos aleatorizados a talla adulta publicados hasta la fecha.
Los estudios publicados son difíciles de comparar debido a la heterogeneidad de diseños, edades, dosis y duración de tratamiento. En general muestran una ganancia significativa de talla adulta de 1,4 ± 0,8 desviaciones estándar (correspondiente a 9,5 ± 5,4 cm)35-38, si bien algunos estudios obtienen ganancias bastante más modestas (tabla 5). En la mayoría de estudios se observa mejor resultado a mayor duración del tratamiento durante la etapa prepuberal. En línea con la hipótesis de que el deterioro del crecimiento en el SN se debe a una resistencia a la hormona de crecimiento, algunos estudios han documentado una pobre respuesta en pacientes con mutación en PTPN1135; sin embargo, otros estudios no han conseguido replicar este resultado37. Aunque el déficit de hormona de crecimiento se ha descrito en algunos pacientes con SN no parece que esta sea la causa fundamental del deterioro de la talla en estos pacientes, con la única excepción de aquellos con mutación en SHOC2, en los que se ha descrito con frecuencia este déficit hormonal. En cuanto a la dosis, estudios recientes muestran una mejor respuesta a corto plazo con dosis más altas37, aunque no hay datos a largo plazo de seguridad y eficacia. En síntesis, los datos disponibles en la literatura médica hacen razonable atender las siguientes recomendaciones:
- -
Independientemente de las condiciones asociadas a la indicación una vez esta se apruebe, el tratamiento con rhGH en pacientes con SN debería ser considerado de manera individual. Deberá tenerse en cuenta la talla, la edad (preferible un inicio precoz para maximizar la ganancia de talla prepuberal), patología asociada (la miocardiopatía hipertrófica no es una contraindicación, pero requiere seguimiento cardiológico estrecho; la escoliosis requerirá seguimiento cercano porque podría empeorar durante el tratamiento; la nutrición debe ser abordada y las deficiencias calóricas resueltas antes de iniciar un tratamiento, si hay datos clínicos de déficit de hormona de crecimiento puede estar indicado estudiar el eje somatotropo) y el genotipo (la indicación en pacientes con variantes de alto riesgo oncogénico debe ser evaluada con especial cuidado y de iniciarse es recomendable un seguimiento estrecho).
- -
La dosis inicial recomendada es de 33 μg/kg/d (si la respuesta no es apropiada podrá incrementarse hasta un máximo de 66 μg/kg/d), con especial atención a niveles de IGF1, metabolismo hidrocarbonado y posibilidad de otros efectos adversos. Si la respuesta no es apropiada tras 1-2 años de tratamiento a pesar de dosis elevadas, considerar suspenderlo, dado que la respuesta máxima debe ocurrir en los primeros años de tratamiento. Los pacientes con diagnóstico clínico confirmado con estudio genético negativo deberían ser candidatos a recibir tratamiento, aunque en esos casos sería deseable disponer de la evaluación por parte de un genetista clínico con experiencia en rasopatías y mantener una actitud crítica a lo largo del seguimiento.
Estudios con hormona de crecimiento en el síndrome de Noonan a talla adulta (adaptado de Carcavilla 2014)
| Referencia | Diseño | Estudio genético | Duración (años) | Edad inicio (años) | HSDS inicio | Dosis(μg/kg/d) | ΔHSDS 1a | Edad/criterio talla adulta | Talla adulta | Δ Talla adulta |
|---|---|---|---|---|---|---|---|---|---|---|
| Kirk 2001 | Observacional KIGS | No | 5,3 | 10,2 ± 3,3 | -2,9 ± 0,7 | 40 ± 20 | 0,4 | ♂ > 17 años♀ > 15 añosVC< 2,5 cm/año | (n = 10)♂159,9 cm(-2,2 SDS)♀147,2 cm(-2,5 SDS) | 3,1 cm(0,8 SDS) |
| Osio 2005 | Ensayo clínico no controlado | No | 7,5 | 7,78,6 | -3,1 ± 0,5-2,7 ± 0,4 | 3366 | 0,8 ± 0,4 | 19,5 ± 1,5 años17 ± 1,4 añosVC < 1cm/año | (n = 18)♂174,5 ± 7,8 cm(-0,9 ± 1,2 SDS)♀157,7 ± 4,7 cm (-1,6 ± 0,8 SDS) | ♂13 cm(1,8 ± 1,0 SDS)♀9,8 cm(1,5 ± 0,8 SDS) |
| Raaijmakers 2008 | ObservacionalKIGS | No | 7,5 | 10,17 | -3,24 | 34(0,17-0,77) | 0,54 | ♂ > 17 años♀ > 15 añosVC< 2 cm/año | (n = 24)ND | 0,61 SDS |
| Noordam 2008 | Ensayo clínico no controlado | PTPN11 (22) SOS1 (1)BRAF (1) | 6,4 | 11 | -2,8 | 50 | 0,5 | 1 año tras suspender GHVC < 2 cm/año | (n = 29)♂171,3 cm♀151,3 cmmedia total:-1,5 SDS | 1,3 SDS |
| Romano 2009 | ObservacionalNCGS | No | 5,6 ± 2,6 | 11,6 ± 3 | -3,5 ± 1♂-3,2♀ -3,8 | 33 ± 5 | ND | ♂: EO ≥ 16♀: EO ≥ 14(proyectada en el 30%) | (n = 64)♂-2♀ -2,3media total:-2,1 ± 1 | 1,4 SDS♂8,9 cm (1,2 SDS)♀ 10 cm (1,5 SDS) |
| Tamburrino 2015 | Ensayo clínico no controlado | PTPN11 (23), RAF1 (1), KRAS (1), SHOC2 (7) | 9,3 ± 4 | 6,9 ± 3,6 | -2,8 ± 0,8 | 35 | 0,53 | VC < 1 cm/año y cierre epifisario | (n = 16)-2,2 ± 0,7 | 0,61 SDS |
| Malaquias 2019 | Estudio longitudinal retrospectivo | PTPN11 (15)Others (2) | 5,1 ± 2,0 | 11,4 ± 3,4 | -3,4 ± 0,8 | 47 | 0,4 ± 0,3 | VC < 1 cm/año durante 12 meses | (n = 17)♂160 cm (13)♀153,5 cm (4) (-2,1 ± 0,7 SDS) (17) | 1,3 ± 0,7 SDS |
| Ranke 2019 | Observacional KIGS | PTPN11 (36) | 6,6 ± 2,8 | 10,4 ± 3,1 | –3,5 ± 1,0 | 36 ± 11 | ND | VC < 2 cm/año y♂: EO≥16♀: EO≥14 | (n = 140)♂ –2,1 ± 1,2 (74)♀ –2,5 ± 1,2 (66)–2,3 ± 1,2 (140) | 1,3 ± 0,7 SDS♂ 1,4 ± 0,8♀ 1,1 ± 0,7 |
HSDS: talla en desviaciones estándar para la población de referencia. HSDS 1 a: Talla al final del primer año de tratamiento en desviaciones estándar para la población de referencia. n: Número de pacientes evaluados con talla adulta o talla próxima a talla adulta. Δ talla adulta: Ganancia media de talla al llegar a talla adulta, calculada como desviaciones estándar para la población de referencia y su equivalente en centímetros. ND: No disponible.
Las estrategias encaminadas a reducir la actividad de la vía RAS-MAPK han recibido una atención considerable con el desarrollo de estudios preclínicos con resultados favorables. Se ha documentado que el uso de trametinib, fuera de ficha técnica, en dos pacientes con miocardiopatía hipertrófica grave que estaban en lista de trasplante consiguió revertir casi completamente la afectación miocárdica39. La inhibición de la vía PI3/AKT/mTOR con rapamicina ha demostrado mejorar la afectación cardiaca en modelos animales de SNLM y documentado su eficacia en un paciente en situación crítica40.
Se ha sugerido también un posible papel de los inhibidores de la 3-hidroxi-3-metilglutaril coenzima A (estatinas) debido a que reducen la farnesilación de RAS y su localización en la membrana plasmática. En un modelo animal de SN el uso de estatinas ha mejorado el crecimiento mejorando la diferenciación de los condrocitos11, y actualmente se está desarrollando un ensayo clínico fase 3 para evaluar la eficacia del tratamiento con estatinas en el crecimiento en el SN (ClinicalTrials.gov identifier NCT02713945).
ConclusionesEl SN es una entidad con una importante variabilidad clínica y genética, que requiere de una aproximación multidisciplinar y un seguimiento regular. El presente trabajo ofrece unas recomendaciones de seguimiento basadas en la información disponible en la literatura en el momento actual. La reciente aprobación por la AEMPS del uso de rhGH en el paciente con SN con talla baja ofrece una herramienta para el tratamiento sintomático no disponible hasta la fecha en nuestro medio.
El desarrollo de los estudios genotípicos ha permitido en las últimas décadas identificar la causa del SN en la mayoría de los pacientes, y ha propiciado el desarrollo de múltiples estudios preclínicos orientados a identificar dianas terapéuticas etiológicas en estos pacientes. Se espera que estos estudios permitan desarrollar en los próximos años tratamientos dirigidos a la raíz de las manifestaciones del SN y el resto de rasopatías.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

