



Introducción
El síndrome de Möbius se considera un defecto del desarrollo del romboencéfalo de gravedad variable1. En una minoría de casos se acompaña de una disfunción respiratoria primaria incompatible con la vida independiente, debido al desarrollo anómalo de la porción dorsal del tegmento pontino caudal. En esta región se encuentran los centros generadores del impulso respiratorio, en la vecindad inmediata de los núcleos de los pares craneales VI y VII2. Este subgrupo de pacientes con síndrome de Möbius presenta riesgo de apneas centrales recurrentes que pueden presentarse en cualquier momento de la infancia1,3. Se presenta un paciente con síndrome de Möbius que desarrolló un episodio aparentemente letal y nos proponemos, a través de nuestros hallazgos, ayudar a identificar pacientes con este síndrome que tienen riesgo de apnea.
Observación clínica
Niño de 7 meses de vida remitido a la unidad de cuidados intensivos pediátricos (UCIP) de nuestro hospital desde otro centro donde había ingresado por un episodio de cianosis, hipotonía y ausencia de respuesta a estímulos mientras dormía al amanecer. A su llegada al servicio de urgencias de dicho centro se realizaron maniobras de reanimación cardiopulmonar avanzada, precisando ventilación mecánica que no pudo ser retirada a partir de ese momento, por lo que fue trasladado a nuestra UCIP.
En la anamnesis destacaban dificultades para alimentar al niño por succión débil y episodios ocasionales de atragantamiento durante los primeros 3 meses, que evolucionaron de manera favorable, junto a ausencia de risa y de llanto desde el nacimiento. Hijo de una madre de 16 años, primípara, primigesta, sin enfermedades previas, cuya exploración no mostraba debilidad muscular ni fenómeno miotónico. No existían antecedentes familiares de enfermedades neuromusculares. Nacido tras un embarazo controlado que cursó con placenta de implantación baja durante el primer trimestre de gestación. La madre había comenzado a apreciar los primeros movimientos fetales al cuarto o quinto mes gestacional. Parto eutócico a las 42 semanas. No precisó reanimación ni ingreso en el período neonatal. Al mes de vida ingresó por infección urinaria. Comenzó la fijación y el seguimiento visual a partir de ese momento, pero nunca se había llegado a observar una sonrisa social. Comenzó a vocalizar, a coger objetos con ambas manos y a darse la vuelta en decúbito a los 4 meses. No había adquirido la sedestación. No tenía antecedentes de otros problemas respiratorios previos.
La exploración física mostraba un lactante sin alteraciones fenotípicas ni manchas cutáneas, conectado a ventilación mecánica a través de una cánula de traqueotomía, que realizaba algunas respiraciones espontáneas, con un nivel de conciencia normal y que mantenía buen contacto visual en la mirada de frente. Presentaba un estrabismo convergente, junto a incapacidad para seguir con la mirada hacia los lados por defecto de la abducción de ambos ojos. La facies era hipomímica con debilidad de la musculatura facial bilateral, más acentuada en la mitad superior de la cara. Realizaba algunas muecas con la musculatura peribucal, pero sin ser capaz de sonreír, reír ni llorar. Tanto la succión como la deglución estaban preservadas, lo que le permitía alimentarse por la boca, en ese momento, sin problemas. Destacaba una tolerancia sorprendente a maniobras dolorosas, que no le ocasionaban apenas ningún cambio en la expresión facial ni sintomatología autonómica acompañante. Junto a ello presentaba una hipotonía no paralítica axial y de extremidades, no mantenía la sedestación y el control cefálico era aún pobre. Los reflejos miotáticos estaban disminuidos en miembros inferiores y bien preservados en los superiores. El fondo de ojo mostraba unas papilas ópticas de aspecto pálido de bordes definidos.
Las exploraciones complementarias realizadas fueron normales: hemograma, bioquímica sérica elemental, creatinfosfocinasa, ácido láctico, piruvato, aminoácidos en suero, ácidos orgánicos en orina, ácidos grasos libres en plasma, carnitina sérica total, carnitina libre, acilcarnitina, cociente acil/libre, equilibrio acidobásico, amonio, hormonas tiroideas, cariotipo, citoquímica de líquido cefalorraquídeo, electromiograma y velocidad de conducción motora de nervio peroneo, biopsia de músculo con estudio estructural, histoquímico y de cuantificación de enzimas de la cadena respiratoria mitocondrial, estudio del ADN mitocondrial de músculo en búsqueda de las mutaciones T8993G, T8993C y A9176C, deleciones únicas o múltiples y depleción por Southern blot, test de edrofonio, estudio de potenciales evocados auditivos del tronco cerebral y una tomografía computarizada (TC) craneal (fig. 1). Se realizaron tres estudios de resonancia magnética (RM) craneal a los 7, 9 y 15 meses de vida que mostraron alteración en la intensidad de la señal en tegmento mesencefálico, pontino y bulbar, más marcado en regiones dorsales (figs. 2 y 3).
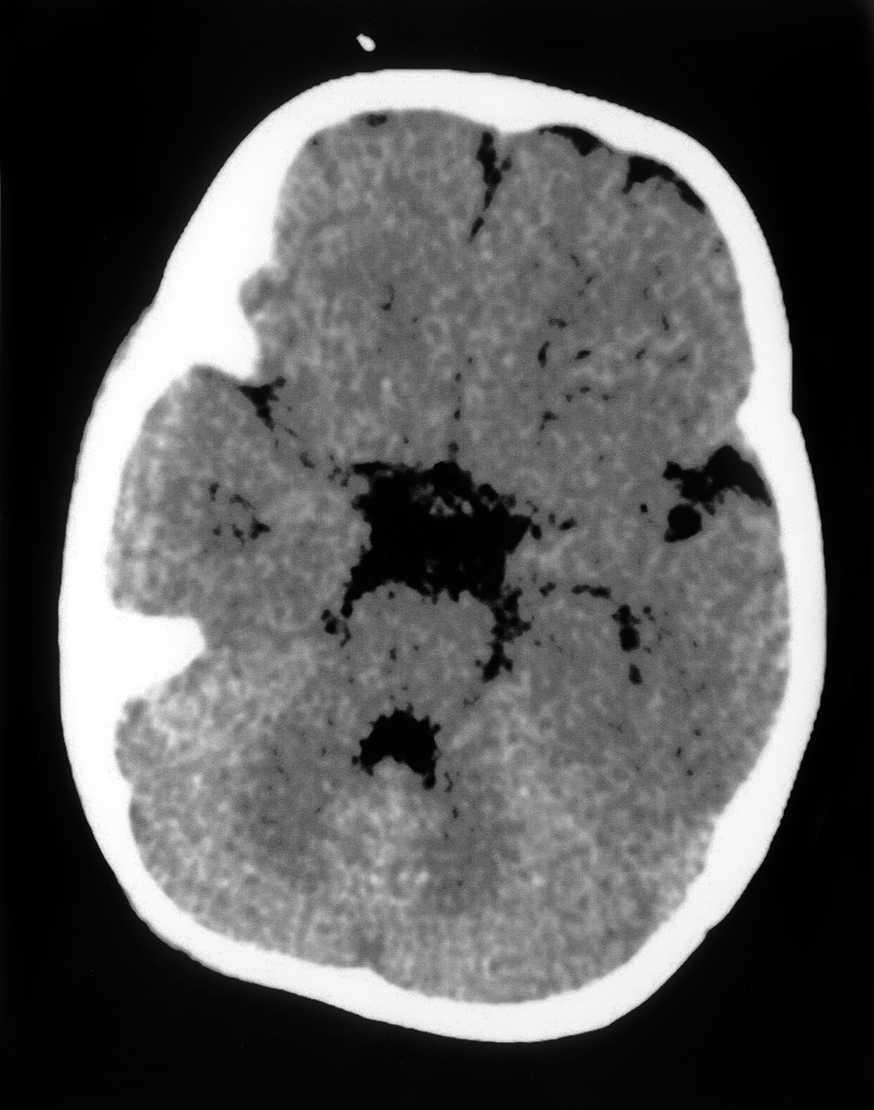
Figura 1.TC craneal a nivel del IV ventrículo: ausencia de hallazgos patológicos.
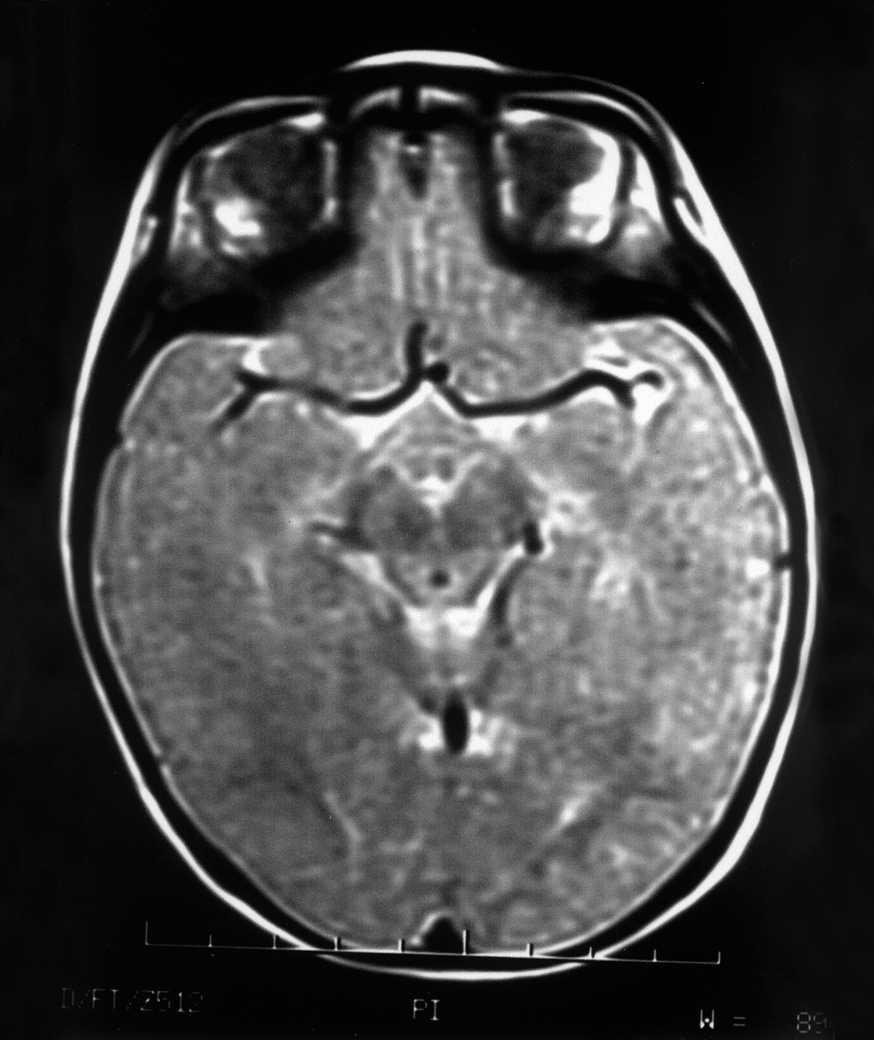
Figura 2.RM craneal: secuencia ponderada en T2 a nivel del polígono. Hiperintensidad de la señal del tegmento paramediano y sustancia gris periacueductal.
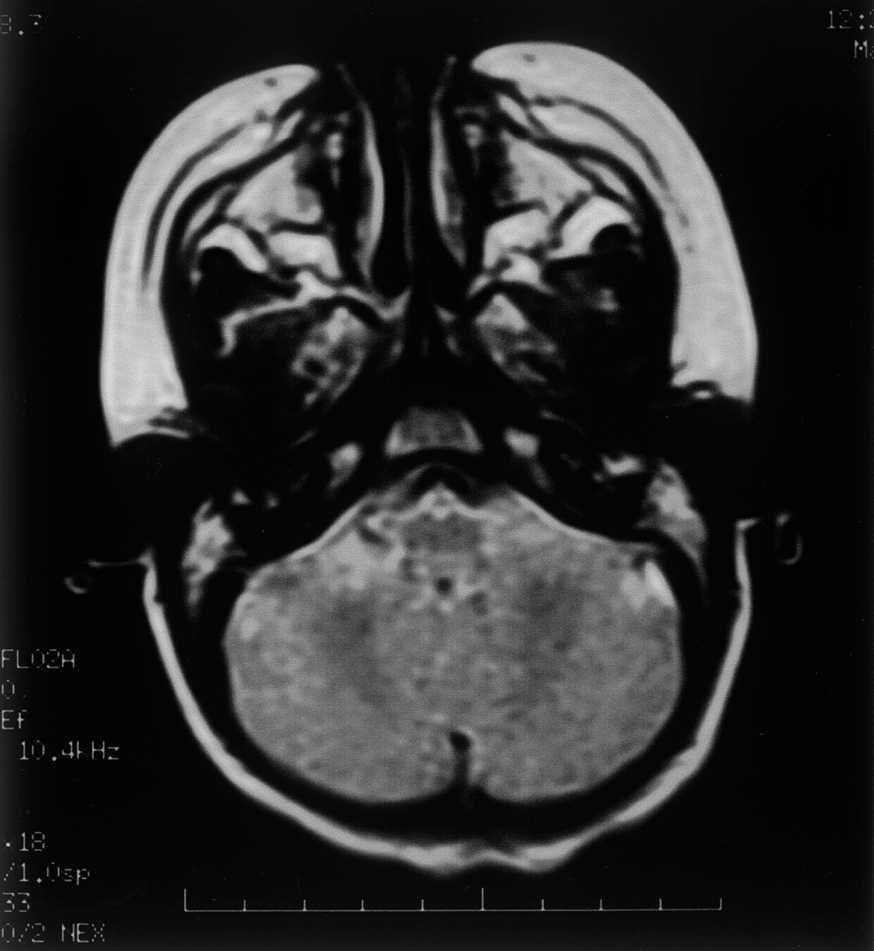
Figura 3.RM craneal: secuencia ponderada en T2 a nivel bulbar. Hiperintensidad de la señal del tegmento paramediano bulbar.
A los 11 meses de vida fue dado de alta con ventilación mecánica domiciliaria, siguiendo controles mensuales en la UCIP. En ese momento precisaba ventilación asistida durante el sueño, y 1 h de cada 3 a 4 h en vigilia. Cinco meses más tarde el paciente falleció en otro centro hospitalario en el curso de un proceso séptico.
Discusión
El síndrome de Möbius se define por debilidad facial congénita junto a afectación de la abducción ocular, aunque algunos autores consideran la diplejía facial congénita aislada suficiente para su diagnóstico. Puede asociar disfunción de otros pares craneales, malformaciones orofaciales y de miembros, defectos musculosqueléticos y retraso mental.
La etiopatogenia del síndrome no está aclarada. Se han propuesto dos orígenes básicos: uno genético4 y otro primariamente vascular isquémico5,6. En ambos se ha considerado la interacción de diferentes teratógenos en la etiología7. Se describe por lo habitual como una condición esporádica, aunque se han publicado casos de herencia autosómica dominante8, autosómica recesiva9 y ligada al X10. También se considera posible su origen poligénico, con factores externos (cocaína, prostaglandinas, talidomida) que podrían desempeñar un papel modificador en las manifestaciones del síndrome1. Las diferentes observaciones anatomopatológicas han sido limitadas en el número de casos y han incluido: agenesia o hipoplasia de los núcleos de los nervios craneales, atrofia de los núcleos de los nervios craneales secundaria a afectación de nervio periférico, necrosis focal del tronco del encéfalo y afectación muscular primaria sin anomalías del tronco del encéfalo ni de los nervios craneales11. Es posible que tanto la hipoplasia nuclear como la necrosis nuclear constituyan un espectro de la misma enfermedad, determinándose la presencia de una u otra por el momento en que se produce el daño durante la gestación, de manera que durante el primer trimestre el tronco del encéfalo respondería a la agresión con cambios malformativos como hipoplasia de los núcleos de los nervios craneales y no sería capaz de responder con necrosis y gliosis hasta el final del segundo trimestre12. Se ha propuesto su clasificación como secuencia malformativa, que se puede presentar aislada o formando parte de cuadros polimalformativos, así como en diferentes síndromes de causa genética, cromosómica o ambiental, haciendo un llamamiento para que deje de utilizarse el término síndrome de Möbius por ser éste generador de confusión, ya que al constituir una secuencia malformativa existe una notable heterogeneidad en las manifestaciones clínicas de los niños afectados, así como en las causas, en el pronóstico y en el riesgo de recurrencia intrafamiliar de esta entidad13.
Existen otras entidades que pueden presentarse con debilidad facial congénita y que deben ser excluidas antes de asumir el diagnóstico de síndrome de Möbius. Entre ellas se incluyen algunas miopatías congénitas, la distrofia muscular congénita, la forma infantil del síndrome facioescapulohumeral, el síndrome de Leigh, la deleción 22q11.2 y otras. En nuestro caso, la biopsia de músculo no reveló alteraciones histopatológicas, bioquímicas ni genéticas, incluido el estudio de la cadena respiratoria mitocondrial y de las mutaciones del síndrome de Leigh de herencia materna. Debido al fallecimiento precoz del paciente no pudo realizarse la determinación de la enzima piruvato deshidrogenasa; sin embargo, consideramos remota la posibilidad de este déficit debido a la normalidad del lactato tanto en suero como en líquido cefalorraquídeo (LCR); la ausencia de otras alteraciones características de esta entidad como signos piramidales, crisis epilépticas, letargia, vómitos, fallo de medro, regresión psicomotora, así como de afectación de los ganglios de la base en la neuroimagen y, por último, la presencia de las manifestaciones clínicas características del síndrome de Möbius desde el primer trimestre de vida sugieren más un defecto estático, de origen prenatal, que una encefalopatía progresiva de origen metabólico. No consideramos necesario excluir la deleción 22q11.2 debido a que la debilidad facial descrita en esta entidad es de distribución unilateral.
El conocimiento del síndrome de Möbius se ha basado en numerosas publicaciones de escasos pacientes, sin embargo, un estudio reciente1 ha incluido a 37 pacientes de diversas edades obteniendo una muestra clínica homogénea cuya etiopatogenia común, sugieren los autores, es un defecto del desarrollo de las porciones caudales del tronco del encéfalo cuya gravedad es variable. En este estudio se describen las características clínicas de estos pacientes, de las que destacamos la alta prevalencia de discapacidades motoras, del 88 % en los pacientes del estudio, incluyendo el 68 % de los pacientes que presentaron retraso motor y un 31 % de pacientes hipotonía importante hasta la edad de 4 años, uno de los signos destacables en el paciente que presentamos. El 34 % de los pacientes mostraban diplejía facial completa, mientras que el 62 % presentaba una relativa preservación de la mitad inferior de la cara, incluyendo los músculos periorales, como también pudimos observar en nuestro caso. Esta mayor afectación de la musculatura facial superior es muy característica de este síndrome y puede tener valor diagnóstico. En 5 de 37 pacientes los padres habían percibido una alta tolerancia de sus hijos al dolor, característica que nosotros también apreciamos. En el 86 % se presentaron dificultades para la alimentación los primeros meses de vida, con un elevado riesgo de aspiraciones recurrentes.
Un subgrupo de pacientes con síndrome de Möbius presenta riesgo de apnea durante toda la infancia, aun sin antecedentes de problemas respiratorios previos. De los 7 niños con alteraciones respiratorias en esta serie1, dos fallecieron por una ausencia del impulso respiratorio presente desde el nacimiento. En 5 pacientes los problemas respiratorios incluyeron: en 2 niños, espasmos del llanto; apneas y bradicardias hasta la edad de 12 semanas en 2 niños y parada respiratoria aguda en otros dos, en un paciente a la edad de un año y en otro a la edad de 13 años. Los 5 pacientes sobrevivieron.
Otros autores han descrito también pacientes con síndrome de Möbius y disfunción respiratoria central2,3,14-16. Igarashi et al3 presentaron 2 pacientes con apneas desde el período neonatal, revisaron la literatura médica disponible hasta 1997, y encontraron otros 13 casos publicados con disfunción respiratoria central. Uno de los dos pacientes continuaba con ventilación mecánica a los 4 meses de vida. El otro paciente fue dado de alta con ventilación mecánica domiciliaria tras 5 meses de hospitalización, y falleció a los 18 meses de vida tras una desconexión accidental del ventilador. Ambos pacientes presentaban calcificaciones en el tegmento del tronco evidenciadas mediante TC craneal. Se realizó RM cerebral en el primero de ellos que no demostró hallazgos patológicos en el tronco.
De los 13 pacientes encontrados en su revisión de la literatura médica, cinco fueron intubados desde el nacimiento debido a ausencia de esfuerzo respiratorio, con una duración del soporte ventilatorio de 65 h a 18 meses. Ninguno de ellos pudo retirarse del respirador. Once de estos 13 niños presentaron apneas desde el período neonatal hasta los 10 meses, algunas de las cuales tuvieron un desenlace fatal. Dos niños que no presentaron apneas mostraban taquipnea continua. Los 13 pacientes habían sido sometidos a estudios de neuroimagen y/o necropsia. En los 7 pacientes en los que se realizó estudio neuropatológico post mortem se encontraron calcificaciones, áreas de gliosis y/o focos de necrosis en el tegmento paramediano del tronco. En 2 niños a los que no se realizó necropsia también se encontraron calcificaciones mediante TC craneal en la misma localización. Estos 9 pacientes tenían antecedentes de apneas. Los 2 pacientes que presentaban taquipnea continua no mostraron calcificaciones en la TC. Únicamente se realizó RM craneal en uno de los 13 pacientes, con antecedentes de apneas, siendo el estudio normal.
Por tanto, 11 de estos 13 casos con síndrome de Möbius y apneas presentaban calcificaciones, necrosis y/o gliosis en el tegmento del tronco, evidentes en TC craneal o necropsia, sin embargo la RM craneal no mostró alteraciones en esa localización en los dos únicos pacientes en que se realizó.
Konkol et al14 comunicaron un paciente con síndrome de Möbius y apneas recurrentes desde el nacimiento hasta los 8 meses de vida. Una TC craneal en el período neonatal mostró dos áreas de hiperatenuación en el puente compatibles con calcificaciones. La RM craneal no mostró hallazgos patológicos. Estos autores sugieren que la TC craneal podría ser más útil que la RM para predecir la disfunción respiratoria central que pueden presentar estos pacientes.
La mayoría de los pacientes con síndrome de Möbius tienen una TC craneal normal17; sin embargo, un estudio reciente describe la presencia de calcificaciones en el tronco en 5 de 7 niños, detectadas mediante TC craneal18. Hay pocos estudios publicados acerca de los hallazgos en la RM craneal de pacientes con síndrome de Möbius. Pedraza et al17 han descrito los hallazgos en 3 pacientes que consistieron en hipoplasia del tronco junto a un enderezamiento del suelo del cuarto ventrículo, consecuencia de la ausencia del colículo del facial o eminencia media. Ninguno de estos pacientes presentaba disfunción respiratoria central asociada. No se encontraron alteraciones en la intensidad de la señal del parénquima del tronco. Estos autores revisaron la literatura médica y encontraron únicamente tres estudios en los que se describían hallazgos en la RM en pacientes con síndrome de Möbius que consistieron en hipoplasia del tronco del encéfalo y en un caso también de cerebelo19-21.
Parece, por lo tanto, claramente definido un subgrupo de pacientes con síndrome de Möbius y riesgo de apnea en los que hay una alta prevalencia de necrosis y/o gliosis junto a calcificaciones del tegmento del tronco, que hasta ahora han sido demostradas mediante TC o necropsia. Este grupo de pacientes representaría los casos con mayor afectación de esta región del rombencéfalo. En el caso que se describe no se encontraron calcificaciones en la TC craneal, pero la RM craneal sí mostró una alteración en la intensidad de la señal en el tegmento paramediano del tronco del encéfalo. Consideramos que la presencia de este hallazgo en la RM craneal de un paciente con síndrome de Möbius puede ser un indicador de riesgo de apnea.
