

El retraso mental (RM) aislado o asociado a otras malformaciones afecta al 2-3% de la población general. Muchas de las causas que originan RM todavía se desconocen. Con los métodos de estudio actuales se puede llegar a identificar la etiología en, aproximadamente, el 50% de los pacientes afectados1-3.
Las anomalías cromosómicas representan un grupo importante entre las causas de RM de base genética, de forma que, por ejemplo, las reorganizaciones subteloméricas son causantes de aproximadamente el 5% de los casos3.
La monosomía 1p36 es el síndrome de microdeleción terminal (no visible mediante cariotipo convencional) más frecuente y por ello mejor caracterizado con una incidencia de aproximadamente 1 cada 5.000 nacidos vivos, y puede determinar entre el 0,5-1,2% de los RM de origen idiopático4-7.
Presentamos el caso de un niño diagnosticado a los 10 meses de edad de síndrome de deleción 1p36, con el objetivo de dar a conocer este síndrome entre los pediatras y otros profesionales para ayudar a orientar su diagnóstico y evitar la realización de exploraciones complementarias innecesarias (como cribado de metabolopatías, neuroimagen, etc.), orientar mejor el pronóstico, ofrecer asesoramiento genético adecuado y proponer un seguimiento apropiado.
Recién nacido varón, de padres no consanguíneos, procedentes de Filipinas. Gestación controlada con detección prenatal de ectasia piélica izquierda. Parto eutócico a las 37,4 semanas, con un peso de 2.970g (− 0,18 DE), talla de 48cm (– 0,6 DE) y perímetro craneal de 34cm (– 0,18 DE). A la exploración neonatal presenta hipotonía generalizada y soplo cardíaco. La ecocardiografía muestra foramen oval permeable, persistencia del ductus arterial y comunicación interventricular membranosa con hipertensión pulmonar severa. El ductus requiere cierre quirúrgico a los 3 meses de edad y posterior tratamiento farmacológico con digoxina durante 3 meses y diuréticos hasta la actualidad. La ecografía renal confirma la dilatación que ha desaparecido en controles posteriores.
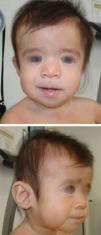
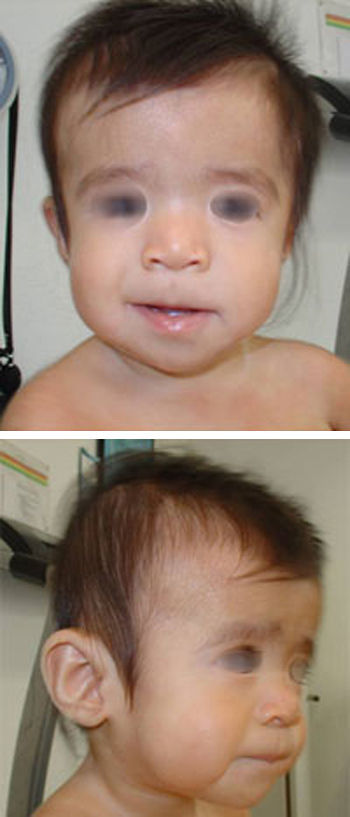
La primera visita por neurología pediátrica a los 7 meses de edad pone de manifiesto estancamiento ponderal y del perímetro cefálico (43cm; – 1,23 DE) con fontanelas amplias (anterior de 8 x 8cm y posterior de 3 x 3cm), retraso del desarrollo psicomotor, hipotonía generalizada y rasgos faciales particulares. Ante la sospecha de cuadro sindrómico, se solicita visita con genética clínica. En la valoración dismorfológica (fig. 1) destacan frente abombada, hipertelorismo, hendiduras palpebrales de inclinación descendente, pliegue epicántico inverso, hipoplasia mediofacial, puente nasal plano, labio superior fino y mentón en punta. Tórax, abdomen, genitales y extremidades, sin anomalías llamativas.
El conjunto de las características orienta hacia una posible cromosomopatía, por lo que se realizan cariotipo que resulta normal (46, XY) y MLPA (multiplex ligation-dependent probe amplification) de regiones subteloméricas (kit comercial de MRC-Holland P036), donde se aprecia deleción 1p36, de la región terminal o subtelomérica del brazo corto del cromosoma 1, descartada en los padres mediante la misma prueba, por lo tanto, de novo.
Se realizó seguimiento del paciente, iniciando estimulación precoz y realizando las siguientes exploraciones complementarias para descartar las anomalías asociadas más frecuentemente: ecografía transfontanelar sin hallazgos significativos, no se realizó resonancia magnética craneal por precisarse anestesia para su realización, y se consideró que, dada la cardiopatía severa que presentaba, era mayor el riesgo que diagnosticar malformaciones cerebrales asociadas; hormonas tiroideas que se normalizaron tras un hipotiroidismo transitorio en el período neonatal; potenciales evocados auditivos de tronco que fueron normales. En la actualidad, está pendiente de la valoración oftalmológica.
El síndrome de deleción 1p36 se caracteriza por hipotonía, RM y/o RDPM, una serie de rasgos faciales característicos y anomalías estructurales, los más frecuentes de los cuales se muestran en la tabla 14,5,7-9. Algunos de los genes incluidos en la región delecionada se han relacionado con características fenotípicas: SKI con fisura palatina, KCNAB2 con epilepsia, MMP23 con fontanelas amplias y GABRD con la alteración del neurodesarrollo5,7-9.
Principales características clínicas del síndrome de microdeleción 1p36 descritas en la literatura5,9
| Clínica | Porcentaje (%) |
| Rasgos faciales dismórficos | |
| Cejas rectas | 100 |
| Filtro largo | 100 |
| Ojos hundidos | 73-100 |
| Puente/raíz nasal amplios | 67-100 |
| Mentón en punta | 63-100 |
| Hipoplasia mediofacial | 51-100 |
| Fontanela anterior amplia con retraso de cierre | 74-85 |
| Microcefalia | 60-65 |
| Braquicefalia | 54-65 |
| Orejas implantación baja/posteriores/anormales | 40-53 |
| Epicantus | 50 |
| Implantación baja del cabello | 48 |
| Hipertelorismo | 38 |
| Fisuras palpebrales pequeñas | 27 |
| Fisuras inclinación ascendente | 27 |
| Fisuras inclinación descendente | 26 |
| Sinofridia | 21 |
| Fisura palatina | 17 |
| Defectos esqueléticos/extremidades | |
| Braquidactilia/camptodactilia | 80 |
| Pies/manos pequeños | 80 |
| Clinodactilia | 40-63 |
| Anomalías esqueléticas | 41 |
| Anomalías viscerales | 71-75 |
| Defectos cardíacos congénitos (FOP, DAP, def. septal ventricular o auricular, válvula bicúspide, anomalía de Epstein) | |
| Miocardiopatía | 23-31 |
| Anomalías gastrointestinales (estreñimiento, reflujo, úlcera, disconfort) | 4-65 |
| Defectos tracto genitourinario | |
| Anomalías genitales externos (criptorquidia) | 25 |
| Anomalías renales | 22 |
| Hallazgos neurológicos | |
| Anomalías EEG | 100 |
| Hipotonía | 87-95 |
| Anomalías cerebrales (RM/TC) (polimicrogiria, leucoencefalopatía, atrofia, ventrículos prominentes) | 88 |
| Convulsiones | 44-56 |
| Dificultad para la alimentación | 47-77 |
| Disfagia orofaríngea | 49-72 |
| Problemas auditivos (sordera neurosensorial o de conducción) | 28-82 |
| Problemas oculares/visión (hipermetropía, miopía, estrabismo) | 17-67 |
| Inatención visual | 30-64 |
| Hallazgos en el desarrollo | |
| Retraso del desarrollo | 100 |
| Lenguaje (pobre/ausente) | 98-100 |
| RM | 95-100 |
| Retraso del crecimiento | 85 |
| Alteraciones del comportamiento | 47-55 |
| Hallazgos endocrinos | |
| Pubertad precoz | 75 |
| Hipotiroidismo | 20 |
| Obesidad | 15 |
La edad de diagnóstico suele ser en los primeros meses de vida y la mayoría antes de los 10 años. Los métodos diagnósticos son el FISH (fluorescent in situ hybridisation) (dirigido o de cribado de regiones subteloméricas) y las técnicas moleculares como la MLPA. El array-CGH (hibridación genómica comparada) y los microarrays, que analizan con alta resolución más regiones del genoma, son mucho más costosos2,3.
El diagnóstico prenatal es posible utilizando las técnicas referidas arriba ante un feto con anomalías compatibles4,5,10.
El manejo clínico de estos pacientes debe ser multidisciplinar, incluyendo de forma obligada seguimiento neurológico (RDPM y riesgo de convulsiones), cardiológico (cardiopatía congénita) y eventualmente valoración visual y auditiva (mediante audiometría y potenciales evocados de tronco). Es fundamental la estimulación precoz y posteriormente el apoyo logopédico6-8,10, y siempre el seguimiento es individualizado, dependiendo de la clínica presente en cada paciente, para no someter al niño a exploraciones innecesarias.
El riesgo de recurrencia de este síndrome para parejas sanas (con cariotipo normal) es inferior al 1%, igual a la población general, debido a que se suele producir de novo por recombinación homóloga no alélica.
Proponemos que los niños con RDPM o RM, trastorno del crecimiento, tanto por falta como por exceso, rasgos dismórficos, microcefalia con o sin anomalía congénita mayor, que en conjunto son sugerentes de cromosomopatía, sean valorados por parte de genética clínica, ya que esta visita, unida a la aplicación de las nuevas técnicas de citogenética y genética molecular, permite un avance espectacular en el diagnóstico del RM idiopático y evita la realización de exploraciones excesivas.
