


El hipopituitarismo congénito es una alteración muy poco frecuente1. Los neonatos con esta enfermedad, pueden estar asintomáticos al nacimiento o presentar síntomas inespecíficos que incluyen hipoglucemia, letargia, apnea, ictericia, colestasis neonatal, convulsiones, dificultad para la alimentación, hiponatremia con potasio normal, sepsis o shock, y en algunos casos, anomalías anatómicas como la presencia de micropene, testes no descendidos, malformaciones craneofaciales o apariencia sindrómica2.
Las mutaciones en genes implicados en el desarrollo de estadios tempranos de la hipófisis, tienden a resultar en formas sindrómicas de hipopituitarismo en asociación con otros defectos congénitos y anomalías de la línea media3–6.
Presentamos a un recién nacido a término y peso adecuado para la edad gestacional, embarazo de riesgo por diabetes gestacional, sospecha de macrosomía fetal y polihidramnios. Parto por vía vaginal, con puntuación de Apgar en el primer minuto de 1, precisando reanimación con bolsa mascarilla, recuperando signos vitales, pero manteniendo un importante distrés respiratorio, por lo que se decidió intubación endotraqueal en paritorio.
Se traslada a nuestro hospital a los 6 días de vida, por fracaso en varias ocasiones de la extubación programada, junto con coagulopatía e ictericia colestática.
El paciente llega intubado, presentando una exploración física en la que destacaba un fenotipo peculiar con facies tosca, cuello corto y pliegue simiesco bilateral, ictericia grisácea y edema en extremidades. Soplo sistólico II/VI rítmico (comunicación interventricular de 3mm en ecocardiograma), aceptable ventilación y un abdomen blando, con hepatomegalia de 2-3cm. Genitales externos masculinos, micropene < 0,5cm y testes palpables en canal inguinal. La fontanela anterior era normotensa con diámetro máximo normal y tendencia a la hipertonía. Pérdida de un 17% del peso al nacimiento. En los estudios iniciales destacaba una glucosa de 14mg/dl, a pesar de tener aportes de glucosa parenterales de 11mg/kg/min, y una bilirrubina total 27,1mg/dl, (directa 4,1mg/dl, GGT 995 UI/L). Ante los hallazgos encontrados se recogieron muestras biológicas para el estudio de errores innatos del metabolismo y microbiológico (que fueron normales), cariotipo (46 XY) y estudio hormonal: prolactina < 2 ng/ml (3,9-29), cortisol 1,8 μg/dl (5-15), T4L 0,4 ng/dl (0,7-1,8), TSH 1,96 μUI/ml (0,47-5,01), IGF-I < 25 ng/ml (114-490), FSH y LH<1 mU/ml (1-12) y testosterona 0,7 ng/ml (2,54±0,78). Se realizó una resonancia magnética nuclear (RMN) donde se observó una hipoplasia hipofisaria con ausencia de tallo y neurohipófisis ectópica, que confirmó el diagnóstico.
Al segundo día de ingreso se inició tratamiento sustitutivo con hidrocortisona iv (dosis inicial 25mg/m2). A los 7 días se añadió tratamiento con levotiroxina oral, continuando con una dosis de mantenimiento de hidrocortisona oral de 17,5mg/m2, con evolución favorable hasta el alta.
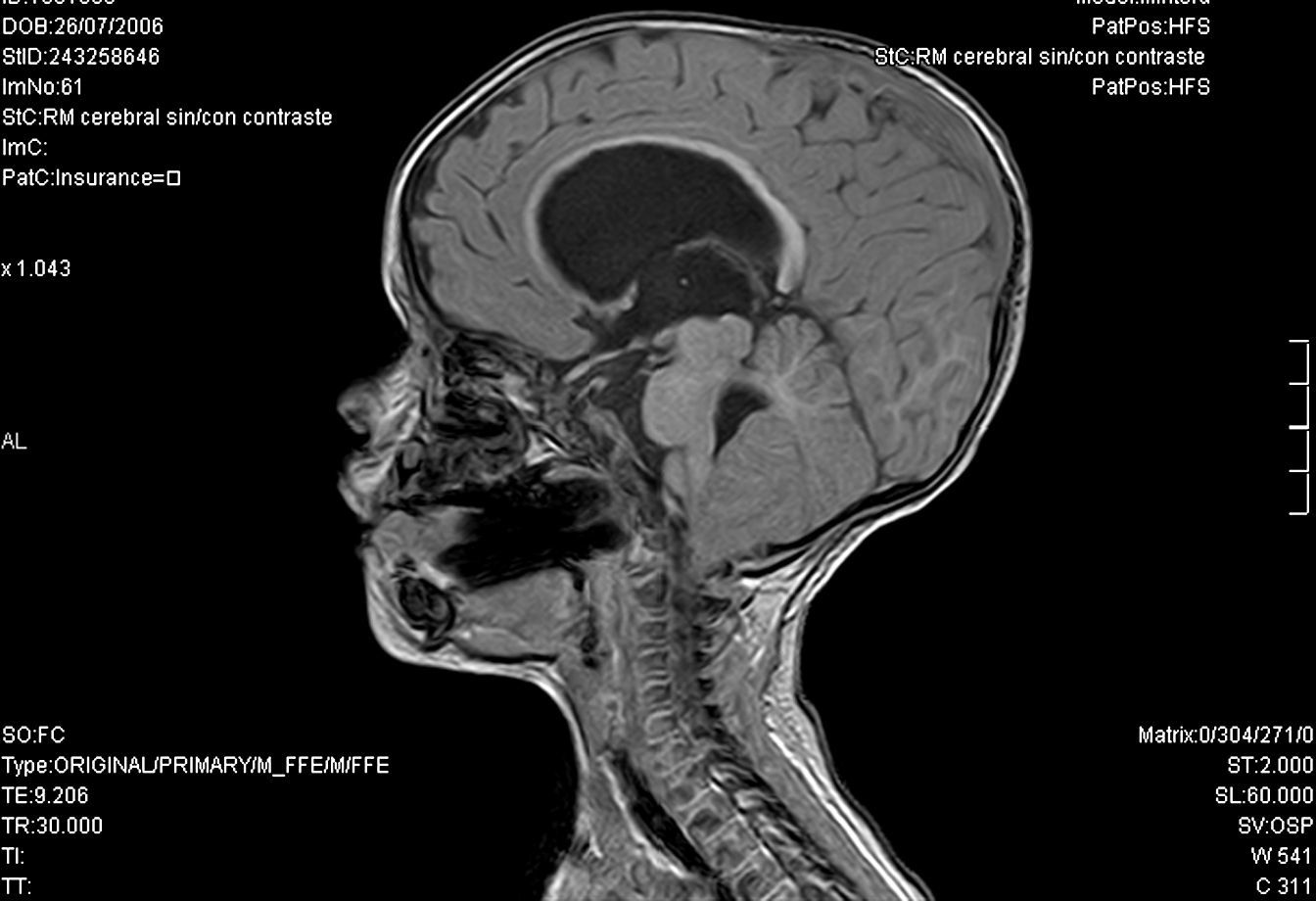
Continuó su seguimiento en consulta con controles clínicos y analíticos dentro de la normalidad, iniciando a los dos meses de vida tratamiento con hormona de crecimiento (0,025mg/kg/día). A los 8 meses de edad, en un estudio de imagen para valorar permeabilidad de coanas se apreció, además de una estenosis de la coana derecha, una dilatación ventricular tetracameral y descenso de las amígdalas cerebelosas, que se confirmó por RNM cerebral (fig. 1). Con carácter de urgencia se colocó una válvula de derivación ventrículo-peritoneal sin incidencias. Durante el postoperatorio inmediato comenzó con poliuria, niveles de sodio plasmáticos en progresivo aumento, y osmolaridad urinaria baja (87 mOsm/L). Ante la sospecha de diabetes insípida se inició tratamiento con desmopresina intranasal con buena tolerancia y control de síntomas.
Al alta presentó buena evolución, seguido en atención temprana por retraso moderado del desarrollo psicomotor y controlado en consulta de neuropediatría. A los 10 meses con el inicio de la dentición, se objetivó la aparición de un incisivo central único en la arcada dentaria superior (fig. 2).
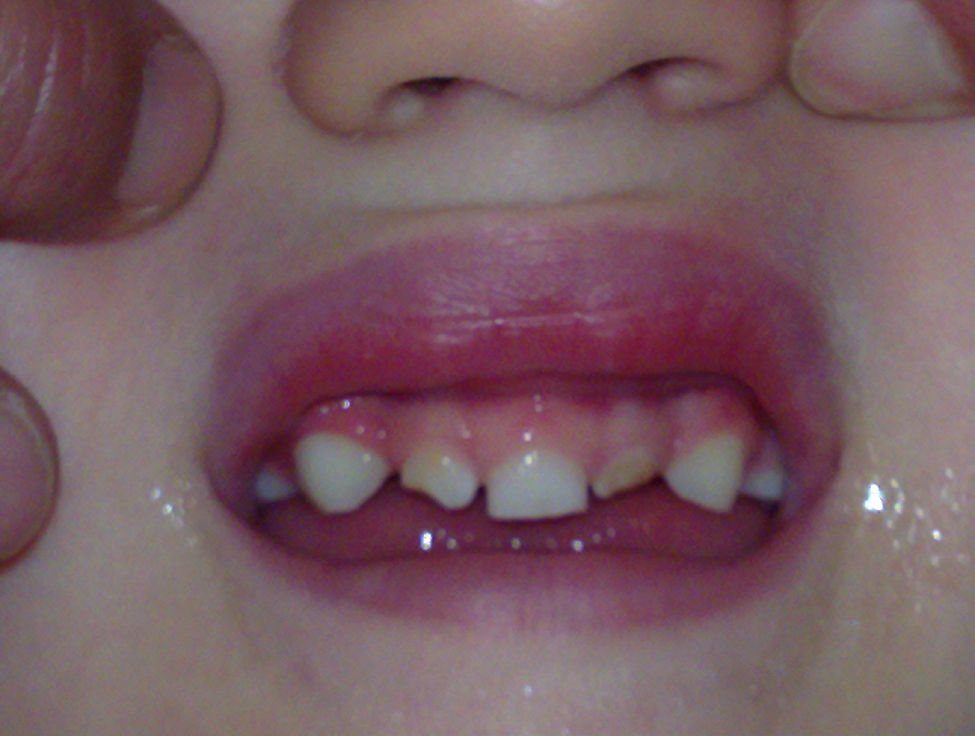
El síndrome del incisivo único central (SIUM) es un error en el desarrollo de la línea media facial muy raro, que puede ocurrir de manera aislada o más frecuentemente como parte del espectro de la holoprosencefalia7. El conjunto de defectos que forman este síndrome son muy variables, desde el espectro completo de la malformación (ciclopia, etmocefalia o cebocefalia), hasta formas más leves como el SIUM8, siendo el gen Sonic Hedgehog Homolog (SHH), el que más frecuentemente se ha descrito asociado al SIUM9.
Se requiere una alto índice de sospecha para el diagnóstico de los neonatos que tienen riesgo de desarrollar hipopituitarismo, ya que hasta un 52% de los pacientes presentarán complicaciones postnatales que pueden amenazar su vida, y el diagnóstico en este período sólo se realiza en un 23% de los casos2. Aquellos pacientes que tengan anomalías oculares, genitourinarias y defectos craneofaciales de línea media, aunque inicialmente puedan estar asintomáticos y las pruebas diagnósticas sean normales, deben ser seguidos a largo plazo porque tienen riesgo de desarrollar con el tiempo un déficit de hormonas hipofisarias2,10.
