






Nuestros objetivos han sido determinar aspectos epidemiológicos, formas clínicas y analíticas de los pacientes con síndrome hemolítico-urémico (SHU) tratados en nuestros centros, así como describir las complicaciones renales y extrarrenales, el tipo de tratamiento requerido y relacionar nuestros casos con la bibliografía actual.
MétodosEfectuamos una revisión retrospectiva de la historia clínica, analítica y estudios de diagnóstico por imagen de los pacientes con diagnóstico de SHU, atendidos desde enero de 1974 hasta agosto de 2007, es decir, durante los últimos 33 años.
ResultadosUn total de 58 pacientes fueron incluidos en nuestro estudio, con una edad media de 2 años y 11 meses; de ellos, estaban ingresados en verano 34 niños. Destaca la presencia de hipocomplementemia familiar en un caso. Con SHU típico (diarrea positivo [D+]) aparecieron 48 casos en los que se aislaron Salmonella enteritidis y Escherichia coli 0157:H7. Con SHU atípico (diarrea negativo [D–]) se contabilizaron 7 casos, y entre las causas destacaban procesos respiratorios de etiología estreptocócica. El recuento plaquetario fue normal en un caso. De los procedimientos empleados cabe destacar que se realizó biopsia renal en 18 pacientes y diálisis peritoneal en 25 casos. Entre las complicaciones se encontraron: en el grupo D+, un caso de necrosis cortical que requirió trasplante renal y en el grupo D–, un paciente con SHU familiar, hipocomplementemia e hipertensión arterial maligna.
ConclusionesHemos llegado a las siguientes conclusiones: a) la enfermedad presenta una baja incidencia en nuestro medio; b) se ha detectado un caso con plaquetas normales; c) un paciente presentó SHU familiar recurrente con hipocomplementemia e hipertensión arterial grave, y d) indicadores como la leucocitosis, la oligoanuria o la hipocomplementemia no influyeron en el curso de la enfermedad.
Our objectives were to determine epidemiology, clinical and laboratory characteristics of patients with haemolytic-uraemic syndrome (HUS) treated in our centre, to describe renal and extra-renal complications and the treatment required and to relate our findings with the existing bibliography.
MethodsWe performed a 33-year retrospective review. We included all patients diagnosed with HUS and monitored in our hospital from January 1974 to August 2007. Clinical histories and imaging studies were reviewed.
ResultsA total of 58 patients were included in our study, with a mean age of 2 years 11 months and most of them were admitted to hospital in summer.
Familial hypocomplementaemia was present in one case. A total of 48 patients presented with typical HUS (with diarrhoea D+ HUS). Salmonella enteritidis and Escherichia coli O157:H7 were isolated from those patients.
While 7 cases presented with atypical HUS (without diarrhoea, D– HUS), most of them associated with a respiratory tract infection due to Streptococcus. In one case platelet count was normal. Kidney biopsy was performed in 18 patients and 25 cases underwent peritoneal dialysis. As regards complications, one child with D+ HUS experienced renal cortical necrosis and required kidney transplant, while in the D-HUS group, the patient with familial hypocomplementaemia had severe hypertension.
Conclusionsa) Incidence of HUS in our environment is low. b) HUS can be present even with a normal platelet count. c) We had one case of HUS in a patient with familial hypocomplementaemia who experienced severe hypertension. d) In our group of patients, the course of the disease was not influenced by the white blood cell counts, decreased diuresis or hypocomplementaemia.
El síndrome hemolítico-urémico (SHU) se define por la presencia simultánea de anemia hemolítica microangiopática (hematíes fragmentados), trombocitopenia e insuficiencia renal aguda1. Es la causa más frecuente de insuficiencia renal aguda en niños con menos de 3 años de edad. Esta entidad puede ser subdividida en SHU típico y atípico según su presentación clínica.
El SHU típico es el más frecuente; se calcula que un 90 % de los casos corresponde a este grupo (diarrea positivo [D+]); se presenta con una fase prodrómica diarreica, a menudo sanguinolenta. La mayoría de los casos se asocian con la bacteria Escherichia coli productora de "Shiga-toxina" (Shiga-liketoxin), también conocida como E. coli productora de verotoxina y E. coli enterohemorrágica (EHEC). Se presenta con más frecuencia en verano.
El SHU atípico es más heterogéneo, y se distingue por la ausencia de diarrea (diarrea negativo [D-]).
La diversidad etiológica del SHU se ve reflejada en numerosos tratados de nefrología pediátrica.
En Argentina, el SHU presenta una incidencia entre 7 y 10 veces superior a la de otras zonas de máximo riesgo. La incidencia anual de la enfermedad en Estados Unidos y en el oeste de Europa es de 2–3 casos por cada 100.000 niños de menos de 5 años de edad1. Los países con mayor incidencia no superan los 3 niños por cada 100.000. Otros países que presentan una relativa frecuencia son Sudáfrica, Países Bajos, País de Gales, Francia y Australia.
E. coli O157:H7 se detecta en el intestino y en las heces. La infección en humanos está provocada por el consumo de carne contaminada poco cocida, leche no pasteurizada o productos lácteos, agua, fruta y vegetales2. La "Shiga-toxina" asociada con el SHU puede contagiar a los hermanos a los pocos días o a las pocas semanas posteriores a la separación del paciente debido a la misma contaminación. Es una enfermedad poco frecuente en neonatos y en niños de etnia negra.
En 1983 nuestro equipo tuvo ocasión de publicar en esta revista una revisión sobre nuestros primeros 9 casos de SHU3. El objetivo de este estudio es revisar los aspectos epidemiológicos, así como las formas clínicas y analíticas de los pacientes con SHU tratados en nuestro centro, así como describir las complicaciones renales y extrarrenales, y el tipo de tratamiento requerido.
PACIENTES Y MÉTODOSHemos efectuado un estudio de observación, longitudinal y descriptivo en todos los pacientes afectados de SHU, 58 casos en total, diagnosticados en el Hospital Infantil Sant Joan de Déu (Barcelona), desde enero de 1974 hasta agosto de 2007.
El diagnóstico se efectuó basándose en la presencia de anemia hemolítica con esquistocitosis, insuficiencia renal aguda y trombocitopenia.
Como SHU típico, asociado a diarreas (D+), se clasificaron 51 casos (87%). En el coprocultivo se aisló Salmonella enteritidis en 6 casos (7%) y E. coli O157:H7 en 4 casos (7%). Los 7 casos restantes se clasificaron como SHU atípico, sin diarrea (D-), constatándose procesos respiratorios en la fase prodrómica, dos de ellos de etiología estreptocócica.
En el estudio se recogen los datos de los pacientes por año, edad de inicio, sexo, mes de aparición, lugar de residencia, así como los gérmenes aislados, aspectos clínicos, biopsia renal en los casos en quienes se efectuó por protocolo, y complicaciones extrarrenales. El tratamiento sustitutivo renal con diálisis peritoneal se efectuó en las siguientes circunstancias: a) oligoanuria que no respondía al tratamiento convencional; b) disminución del filtrado glomerular por debajo de 10ml/min/1,73 m2; c) hiperpotasemia grave que no respondía al tratamiento convencional y con alteraciones en el ECG, y d) hipervolemia grave que comportaba hipertensión arterial y no respondía de forma adecuada al tratamiento hipotensor.
También se ha considerado en el estudio el tipo de tratamiento adoptado, así como el tipo de secuelas derivadas de la enfermedad.
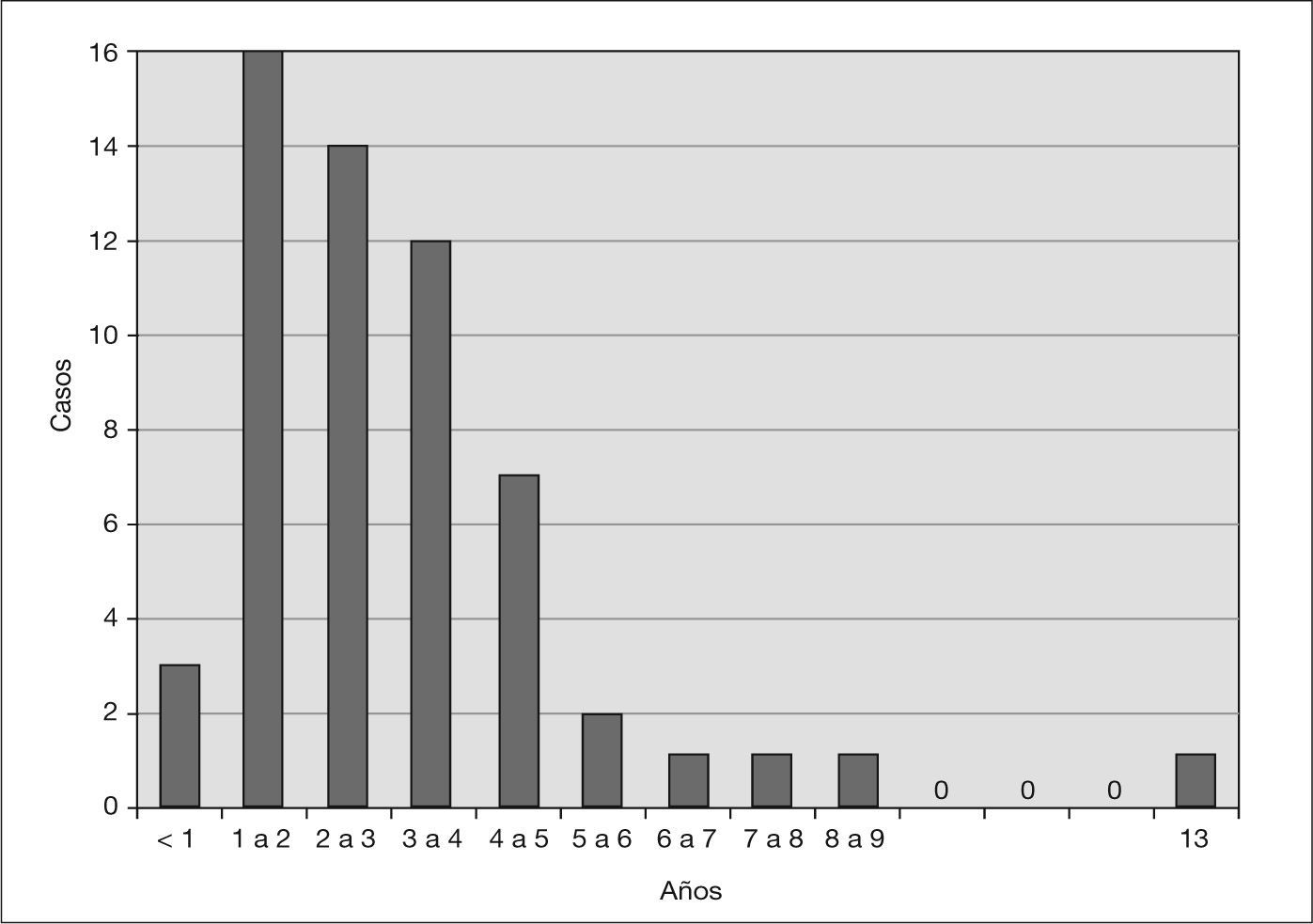
RESULTADOSDe los 58 casos de SHU, 32 (55 %) eran mujeres y 26, varones (45%). La edad media fue de 2 años y 11 meses, con un rango de edad comprendido entre 5 meses y 13 años. El mayor predominio de enfermos se sitúa entre 1 y 3 años (fig. 1).
Entre los antecedentes cabe destacar en un paciente la presencia de hipocomplementemia, al igual que se observó en su padre (fallecido), en una tía paterna y en el hijo de ésta, afectados de SHU. Hoy día este paciente, con 30 años, presenta insuficiencia renal crónica e hipertensión arterial maligna en tratamiento, como secuela de la enfermedad. En otro paciente, la madre era portadora del virus de la inmunodeficiencia humana (VIH), aunque el niño era negativo; la enfermedad no dejó ningún tipo de secuela. En un tercer paciente, con buena evolución de su enfermedad, la madre presentaba un déficit de proteína S. En nuestro último caso destaca la llegada del niño desde Argentina 4 días antes de su ingreso (D+).
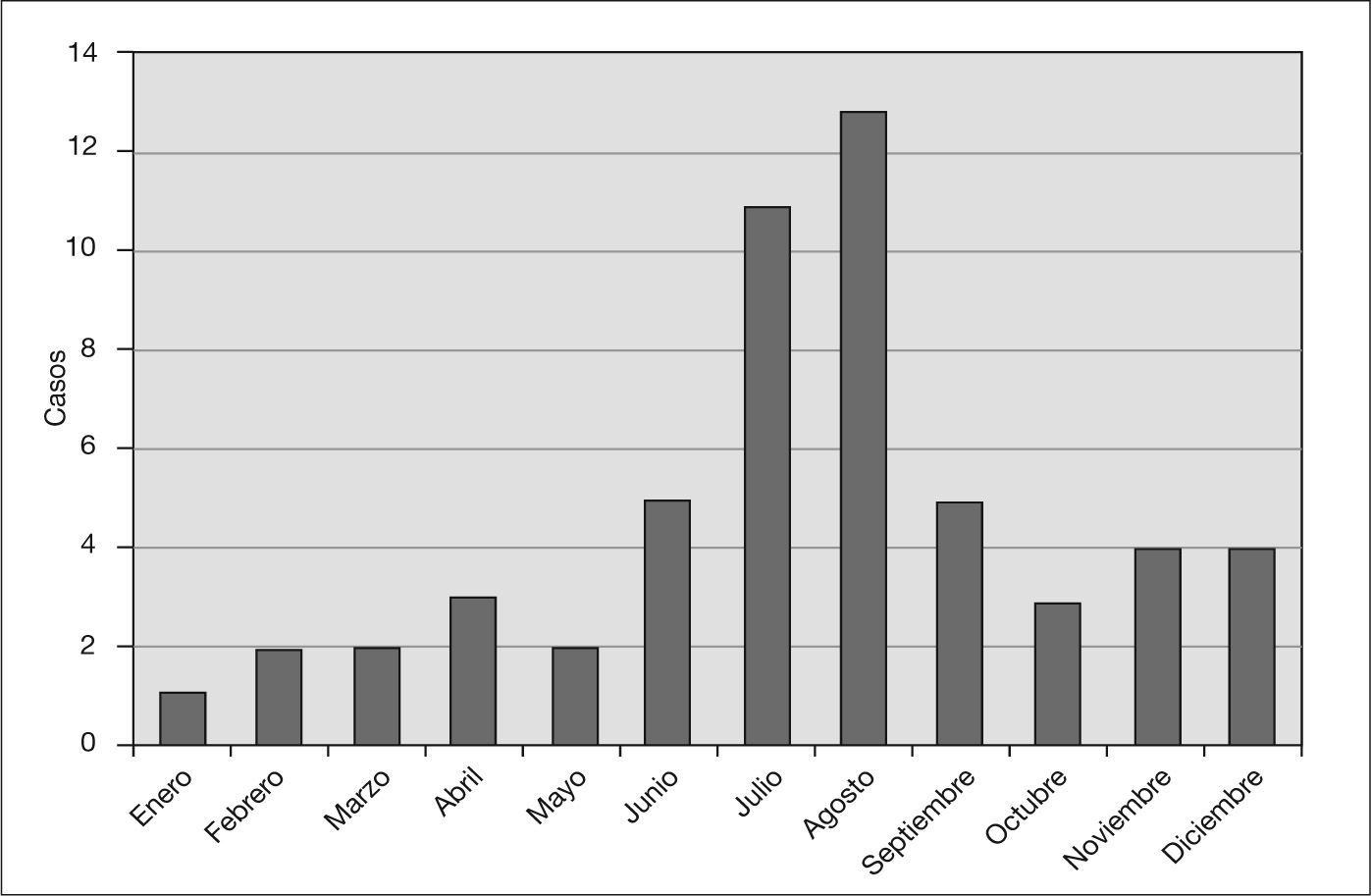
Los meses de mayor predominio de la enfermedad fueron julio y agosto. En el período estival fueron ingresados 37 pacientes, lo que supone un 63% del total (fig. 2). La incidencia de SHU osciló entre uno y cuatro por año. El lugar de procedencia del paciente fue muy variado, con un predominio de Barcelona ciudad y de la comarca del Baix Llobregat, adjunta a la ciudad y de similares condiciones climáticas.
La sintomatología clínica de los pacientes se expone en la tabla 1.
La anemia hemolítica con esquistocitosis se detectó en todos los niños, con hemoglobinas que oscilaron entre 4,8 y 9,7g/% y hematocritos del 12–28,7%. La insuficiencia renal aguda estuvo presente en todos los casos, con cifras de uremia que oscilaron entre 58 y 463mg/ % y cifras de creatinina sérica de 0,9 a 11,8mg/%. La trombocitopenia se constató en casi todos los casos; únicamente en un caso el recuento plaquetario fue normal, aunque con un inicio típico de SHU, y el diagnóstico se confirmó mediante biopsia renal (microangiopatía trombótica glomerular).
La hematuria estuvo presente en 45 casos (77%), microscópica en 39 casos, 37 casos D+ y 2 casos D(86 %), y macroscópica en 6 casos (14%), 4 casos D+ y 2 casos D-. En 45 pacientes (77%) se detectó proteinuria, que en 23 casos se encontraba en el rango nefrótico (51 %), 22 casos D+ y un caso D-, y en 22 casos no alcanzó el rango nefrótico (49%), 19 casos D+ y 3 casos D-.
Se confirmó la existencia de hipertensión arterial en 18 niños (31 %), con una presión arterial sistólica media de 157mmHg y una presión arterial diastólica media de 97,5mmHg; únicamente se constataron 2 casos de encefalopatía hipertensiva, con una presión arterial máxima de 210/150mmHg. Dos casos de hipertensión arterial fueron D–y los casos de encefalopatía hipertensiva fueron D+.
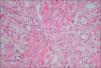
La biopsia renal se efectuó en 18 pacientes (31 %); en 16 casos se diagnosticó una microangiopatía glomerular (MAT glomerular) (fig. 3), en 4 casos D–; un caso de microangiopatía trombótica vascular fue D– (MAT vascular) y un caso de necrosis cortical D+ (fig. 4). En los últimos 12 años, hemos diagnosticado 30 niños en quienes sólo se ha realizado biopsia en tres (D+), lo que representa un 10 % de estos pacientes. El motivo para practicar la biopsia renal fue una oligoanuria de 30 días de duración y el resultado fue MAT glomerular; en el segundo caso, el recuento de plaquetas fue normal y el resultado histopatológico fue también de MAT glomerular, y en el tercer caso, la presencia de una insuficiencia renal crónica terminal fue motivada por necrosis cortical (trasplante renal) (fig. 3).
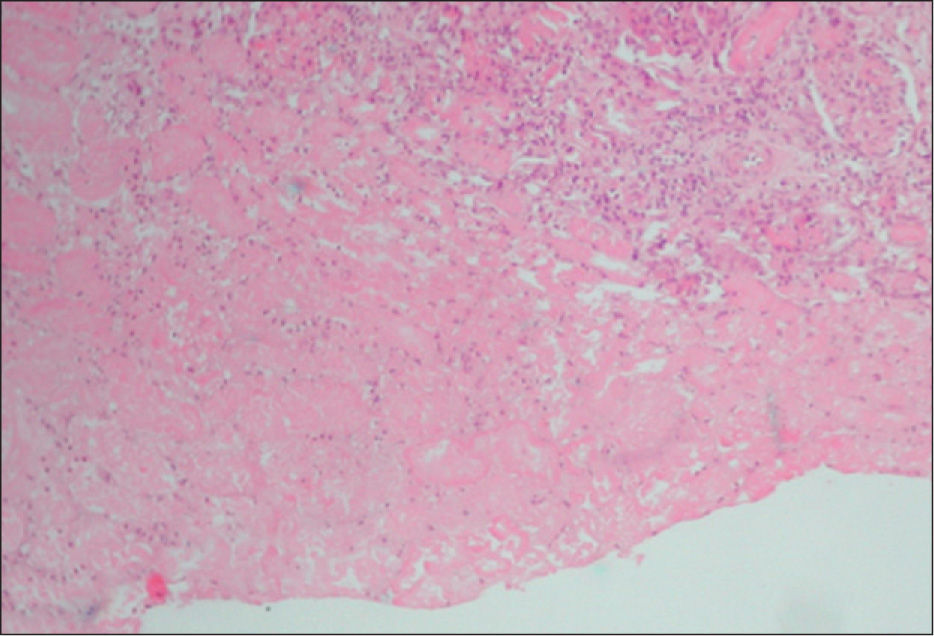
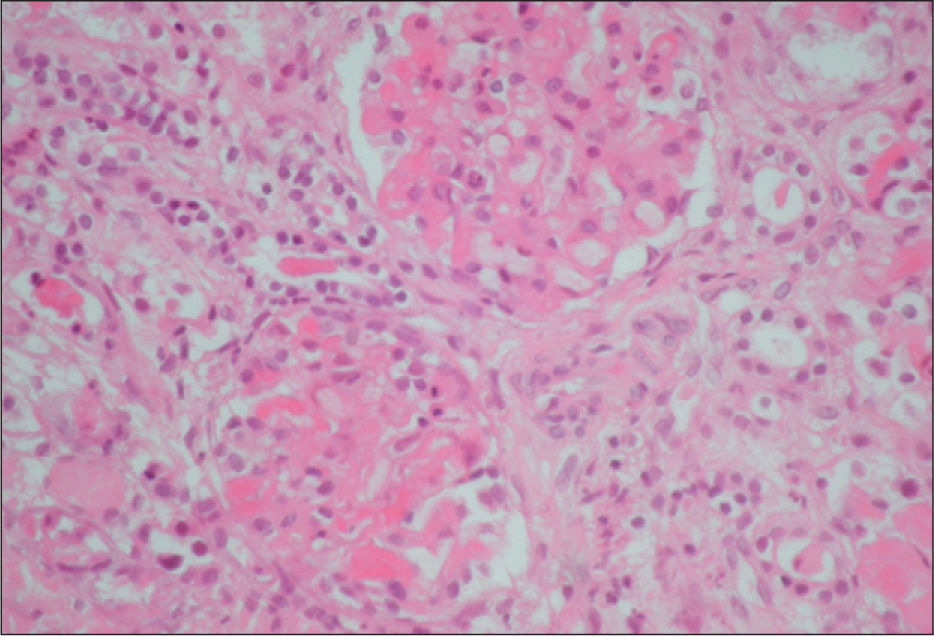
El tratamiento de sustitución renal con diálisis peritoneal se efectuó en 28 casos (48%). De estos pacientes, 2 casos precisaron hemodiálisis transitoria por razones hemodinámicas y un caso necesitó hemodiafiltración.
Se realizaron transfusiones cuando la hemoglobina era inferior a 6g/dl o el hematocrito era inferior al 18%. Se realizaron transfusiones en 56 niños (96%), con un rango de 1 a 6 transfusiones por paciente. Se intentaron mantener concentraciones de hemoglobina comprendidas entre 8 y 9g/dl, para prevenir complicaciones cardíacas y pulmonares. En un paciente se requirió la transfusión de plaquetas porque presentaba una hemorragia digestiva grave. En un paciente se utilizó la eritropoyetina por vía subcutánea con una buena respuesta. Entre los niños en quienes se realizaron transfusiones, cinco fueron D–.
Los fármacos hipotensores utilizados principalmente en los últimos años fueron los antagonistas del calcio (nifedipino) y los inhibidores de la enzima conversiva de la angiotensina (captopril). La encefalopatía hipertensiva se trató con nitroprusiato sódico.
En el apartado de complicaciones se produjo una pancreatitis aguda en 2 niños que no requirieron insulina (D+). El primer caso cursó con oligoanuria de 14 días de duración que precisó tratamiento de sustitución renal con diálisis peritoneal. La proteinuria en el rango nefrótico y la microhematuria desaparecieron a los 4 meses. El tratamiento requirió alimentación parenteral durante 2 semanas. El segundo paciente presentó oligoanuria durante 12 días, que requirió tratamiento con diálisis peritoneal. La microhematuria desapareció al mes. Se instauró también alimentación enteral (por sonda transyeyunal).
En el curso de la enfermedad un paciente presentó una hemorragia digestiva grave que precisó transfusión de plaquetas, con nefropatía mínima, y posteriormente tuvo una buena evolución (D+).
Las complicaciones neurológicas se presentaron en 22 niños (40 %) con algún tipo de sintomatología neurológica inicial, como somnolencia, irritabilidad o crisis comiciales; 5 casos presentaron secuelas neurológicas, que precisaron un control evolutivo durante 12–16 años.
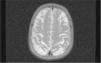
El primer caso D+ (una niña de 14 meses de edad) cursó con convulsiones tónico-clónicas, accidente vascular agudo y hemiparesia. A los 22 meses presentaba un desarrollo dentro de la normalidad salvo por una leve paresia del lado izquierdo. A los 16 años la paciente tiene un examen neurológico normal. El segundo caso D+ presentó disminución del nivel de conciencia, hiperreflexia y falta de respuesta a estímulos, con movimientos oculares horizontales lentos. En el electroencefalograma se detectó una afectación hemisférica bilateral y en la resonancia magnética cerebral se observaron lesiones inflamatorias corticales y subcorticales, así como afectación de los núcleos de la base. Actualmente la paciente presenta dificultades de comprensión y percepción visual (fig. 5).
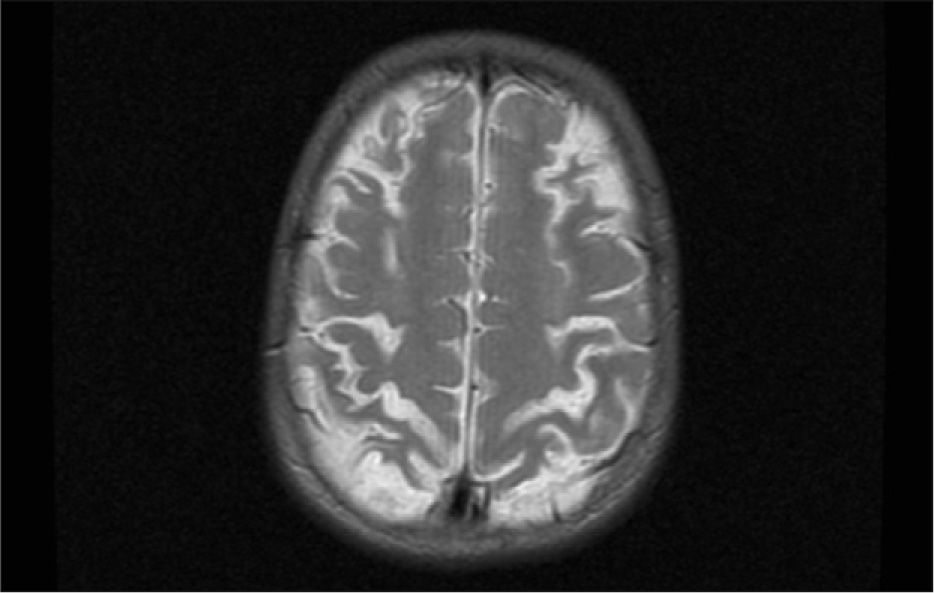
El tercer paciente era una niña de 22 meses, D+, que en las primeras 24h de evolución manifestó un cuadro de miosis con desviación mantenida de la mirada, hipertonía de extremidades inferiores e hiperreflexia generalizada. La niña se recuperó neurológicamente. En los controles clínicos posteriores realizados los 4 años se ha observado un leve retraso en la adquisición de habilidades.
El cuarto caso era un niño de 14 meses de edad (D+); en su segundo día de hospitalización presentó una crisis comicial tónico-clónica generalizada que cedió al administrar diazepam. Tras ser dado de alta no fue traído para realizar sus controles evolutivos. A los 6 años de edad sus padres consultaron por su bajo rendimiento escolar, pero no siguieron los controles posteriores.
El último caso es el de un niño de 3 años y 11 meses (D+), quien, a las 48h del ingreso, inicia un cuadro de irritabilidad con períodos de agitación y de obnubilación. La resonancia magnética craneal fue normal. El niño mejoró paulatinamente en 8 días. Al año del alta, el control neurológico del niño ha sido normal.
DISCUSIÓNSegún nuestra experiencia, la edad media de presentación, la incidencia según el sexo y el aumento de frecuencia en verano coinciden plenamente con los datos referidos en la bibliografía médica4. Los casos por año oscilaron entre uno y cuatro. El SHU nunca se presentó de forma epidémica; 33 casos eran de nuestra área de referencia del hospital.
La mayor frecuencia de SHU típico, o D+, coincide con los datos comunicados por otros autores. En nuestra serie se ha producido una baja incidencia de fase prodrómica provocada por E. coli frente a la indidencia referida en otras series4,5.
La anemia hemolítica y la insuficiencia renal estuvieron presentes en todos los pacientes, mientras que el recuento plaquetario fue normal en un paciente D+, hecho comunicado en la bibliografía médica en un 6 % de los pacientes6.
La anemia hemolítica se ha caracterizado por la presencia de una hemoglobina inferior a 8g/dl, negatividad del test de Coombs y presencia de esquistocitosis (hematíes fragmentados) en sangre periférica. La plaquetopenia se situó por debajo de 60.000/μl. La oligoanuria en la insuficiencia renal aguda estuvo presente en la mitad de los casos. La hipertensión arterial también estuvo presente y deben tenerse en cuenta como causa el exceso de líquidos y las trasfusiones de sangre. La presencia de hematuria fue patente, más frecuente en su forma microscópica. La proteinuria se presentó tanto en forma nefrótica como no nefrótica.
Gerber et al4, en un estudio multicéntrico efectuado conjuntamente en Austria y Alemania, analizan los datos de 394 niños afectados de SHU, con una edad media de 3 años, edad similar a la de nuestros pacientes; el 91 % de los casos tenían un episodio prodrómico diarreico (un 57 % de diarreas hemorrágicas), frente a nuestro porcentaje, que fue del 87 %; se evidenció infección por E. coli en un 83 % de los casos, mientras que en nuestra casuística este porcentaje fue sólo del 7 %. La mitad de los pacientes enfermaron entre junio y septiembre, y en nuestra serie la incidencia fue del 63 %.
El SHU afecta7 al sistema nervioso central (SNC), al sistema digestivo, al sistema cardiovascular, al páncreas y al hígado. La afectación del SNC se presentó en un 20 % de los niños con SHU típico con la presencia de convulsiones, coma, hemorragia cerebral y hemiparesia. La afectación grave del SNC se asocia con una importante mortalidad, aunque en nuestro grupo no hubo mortalidad secundaria de causa neurológica.
El sistema gastrointestinal puede verse afectado en forma de colitis ulcerosa grave, necrosis intestinal, perforación e invaginación intestinal. En un caso tuvimos ocasión de observar una hemorragia digestiva grave y en el segundo caso se observó la presencia de una vasculitis gástrica y una úlcera duodenal hemorrágica.
La afectación cardíaca puede deberse a isquemia, y sus indicadores más fiables son el aumento de troponina 1, la intoxicación urémica y la hipervolemia.
La afectación pancreática, según se refiere en la bibliografía científica consultada, es inferior al 10 %; en nuestros pacientes representó alrededor del 3 %, y ninguno presentó intolerancia a la glucosa o diabetes. La afectación hepática en esta enfermedad es rara, y no hubo ningún caso en nuestra serie.
La afectación histológica renal8 puede ser de tres tipos: a) microangiopatía trombótica (MAT) glomerular, la lesión histológica más benigna; b) necrosis cortical, forma más grave que se observa con mayor frecuencia en el SHU típico y que puede conducir al paciente a insuficiencia renal crónica terminal, y c) MAT, que se observa con mayor frecuencia en el SHU atípico y que puede provocar hipertensión arterial maligna.
En nuestro grupo de enfermos, el único caso de hipertensión arterial maligna, familiar e hipocomplementemia, corresponde a una MAT vascular. En el único caso sometido a trasplante en la biopsia renal se observó necrosis cortical.
E. coli enterohemorrágica (EHEC) difiere de otras E. coli patógenas por la producción de la toxina "shiga", que contribuye a la patogenia y a la evolución del SHU típico. La bacteria más prevalente de este grupo en Estados Unidos es E. coli O157:H79.
Se ha calculado que 75.000 pacientes al año presentan enfermedad por EHEC en Estados Unidos10; de este grupo, un 30% sobrepasa los 50 años. En 2003, E. coli estuvo implicada en un 3 % de los pacientes afectados por enfermedades transmitidas por la alimentación11. Se ha demostrado que el aislamiento de E. coli 0157:H7 es más frecuente en las muestras de heces con sangre que en las heces que no la contienen (el 7,8 frente al 1,4%). El período de incubación entre el contagio de EHEC y el inicio de la enfermedad es de 4 días.
La administración de polímeros del globotriaosilceramida, (Gb3 receptor de la toxina "shiga"), por vía oral, disminuye las concentraciones séricas de la toxina y las lesiones cerebrales ocasionadas por ésta, aunque este hecho ha sido demostrado en animales de experimentación y no en humanos12,13.
El SHU atípico, o D–, se caracteriza por ausencia de fase prodrómica, comienzo insidioso, recaídas frecuentes, hipertensión arterial e insuficiencia renal crónica; puede haber consumo de C3 y presencia de antecedentes familiares14.
En Canadá, aproximadamente el 40 % de los casos D– presenta una infección neumocócica15.
Siegler et al distinguen entre 22 casos de SHU atípico y 265 casos de SHU típico16. En el 6 % de los pacientes con SHU el recuento plaquetario fue normal. En la fase prodrómica de los SHU atípicos se observaron infección respiratoria de vías altas (23%), fiebre no catalogada (46%) y vómitos (81 %). No hubo diferencias en el hematocrito ni en el recuento plaquetario de los dos grupos, pero las cifras de creatinina fueron más bajas en el grupo D– (1,2 frente a 2,2mg/dl). La oliguria apareció en menos casos en el grupo D– (21/47%). No hubo ningún fallecimiento en el grupo D–, mientras que en el otro grupo la mortalidad se situó en el 3,4%. En el seguimiento durante 6 años no se apreciaron diferencias significativas en cuanto a la aparición de hipertensión arterial o proteinuria. La incidencia de la insuficiencia renal crónica fue similar en ambos grupos.
La lesión histológica renal característica del SHU atípico es la microangiopatía trombótica arteriolar, mientras que en el grupo de SHU típico la lesión más frecuente es la microangiopatía trombótica glomerular, hecho que se corresponde con lo observado en nuestra serie.
La asociación de la enfermedad con el VIH se ha referido en la bibliografía17 y destaca que la mayoría de los niños acaban presentando una insuficiencia renal crónica terminal.
El SHU de causa genética está aumentando18 como deficiencia de los componentes del complemento, principalmente la fracción C3, déficit de ADAMTS 13 y defectos intracelulares del metabolismo de la vitamina B12. Anormalidades de los genes del factor H, proteína cofactor de membrana (MCP) y factor I se asocian con el SHU atípico, por lo que el 50 % de los casos de SHU atípico es el resultado de mutaciones de estos genes19.
El SHU asociado con deficiencia del factor H se presenta durante la infancia, pero algunos casos se han observado en adultos20.
Rodríguez de Cordile et al21 han identificado un gen que confiere un riesgo elevado de SHU atípico y que posee las mismas mutaciones en las proteínas reguladoras del complemento. La mutación se ha descrito en la proteína factor B. Este trabajo ofrece buenas perspectivas para el diagnóstico molecular: identificar el gen mutado es un buen indicador pronóstico del trasplante renal.
La prevención de la infección por EHEC mediante protocolos efectuados por expertos en salud pública y veterinaria tendría una influencia importante en la incidencia mundial del SHU22.
En conclusión, el SHU presenta una baja incidencia en nuestro medio. Destaca en nuestra serie la presencia de un paciente con recuento plaquetario normal, y la existencia de un solo caso de SHU familiar recurrente, hipocomplementemia e hipertensión. Los indicadores, como la leucocitosis, oligoanuria e hipocomplementemia, no influyen en el curso de la enfermedad.
