

El síndrome de Guillain-Barré (SGB) es una polineuropatía aguda de difícil diagnóstico en la primera infancia.
ObjetivosRevisar la forma de presentación del SGB en niños menores de 6 años atendiendo al tiempo de evolución y sintomatología que presentaron hasta el diagnóstico, los hallazgos en las pruebas complementarias y la evolución y pronóstico.
Pacientes y métodosRevisamos a todos los pacientes menores de 6 años que cumplieran los criterios de Asbury et al para el diagnóstico de SGB.
ResultadosSe incluyó a 8 pacientes, con una media de edad de 3,4 años. El 75% se registró la presencia de un agente infeccioso previo. La sintomatología previa al diagnóstico fue de carácter muy heterogéneo, lo que conllevó un amplio diagnóstico diferencial y multitud de exploraciones complementarias. El tiempo medio al diagnóstico fue de 8,5 días. El 100% presentó afectación motora de miembros inferiores, el 75% de miembros superiores y el 12% de musculatura respiratoria. La afectación sensitiva fue del 62,5% y la de pares craneales del 25%. Requirieron ingreso en cuidados intensivos un 25%. Se objetivó disociación albúmino-citológica en el líquido cefalorraquídeo (83,3%) y positividad de todos los estudios electrofisiológicos con distintos patrones. El pronóstico fue excelente en todos los pacientes.
ConclusionesEl SGB en niños menores de 6 años es de difícil diagnóstico por la inespecificidad de las primeras manifestaciones en muchas ocasiones. Esto implica un amplio diagnóstico diferencial y retraso diagnóstico. Es relevante, el buen pronóstico en este grupo de edad de todos los subtipos electrofisiológicos.
Guillain-Barré syndrome (GBS) is an acute polyneuropathy that is difficult to diagnose in young children.
ObjectivesTo review the form of presentation of GBS in children under six years-old at the time of onset and the symptoms they had until the diagnosis, the findings in the complementary tests, and the progression and prognosis.
Patients and methodsAll patients less than 6 years-old who fulfilled the Asbury et al criteria for the diagnosis of GBS were reviewed.
ResultsEight patients with a mean age of 3.4years were included. Of those 75% recorded a previous infection. The symptoms prior to the diagnosis were very heterogeneous which entailed a wide differential diagnosis with many complementary examinations. The mean time to diagnosis was 8.5days. All of them (100%) had motor involvement in the lower limbs, 75% in the upper limbs and 12% in the respiratory muscles. Sensory and cranial nerve involvement was observed in 62.5% and 25%, respectively. Admission to intensive care was required for 25% of the patients. Albumino-cytological dissociation was observed in the CSF in 83.3% and all the electrophysiological tests were positive with different patterns. The prognosis was excellent in all patients.
ConclusionsGBS in children under 6 years-old is difficult to diagnosis due to the signs of onset often being unspecific. This entails a wide differential diagnosis, with the subsequent diagnostic delay. There is a good prognosis in all the electrophysiological sub-types in this age group.
El síndrome de Guillain-Barré (SGB) se define como una polineuropatía inflamatoria aguda caracterizada por una parálisis flácida arrefléctica, ascendente y simétrica1–4. Posteriormente, esta definición se ha ido progresivamente ampliando a un gran espectro de polirradiculopatías agudas de características heterogéneas resultantes de respuestas inmunológicas, probablemente desencadenadas por agentes infecciosos contra distintos componentes del nervio periférico (células de Schwann, nódulos de Ranvier, etc.) y con un carácter autolimitado1,5.
De entre ellas se describen: neuropatía desmielinizante inflamatoria aguda, neuropatía motora axonal aguda, neuropatía sensitivomotora axonal aguda4. Sin embargo, existen distintas variantes en la presentación clínica, siendo la más reconocida el síndrome de Miller-Fisher6,7. Afecta a 1 caso por cada 100.000 habitantes/año, con un ligero repunte en la incidencia en adolescentes y ancianos1,8. Son varias las publicaciones que hacen hincapié en las múltiples variantes inusuales con las que se presenta en las edades más precoces de la infancia y en la multitud de exploraciones complementarias que se practican previamente a la confirmación diagnóstica3,4,9,10. Entre ellas, se consideran: formas asimétricas, motoras puras, predominantemente sensitivas o sensitivas puras11, autonómicas puras, asociadas a manifestaciones del sistema nervioso central, con reflejos osteotendinosos normales o incluso aumentados, parálisis descendentes1,4,9,10 y variantes regionales de la forma desmielinizante aguda como la parálisis faringocervicobraquial9–11, polineuropatía craneal múltiple4,12,13, paraparesia arrefléctica4,9,10, paresia del VI par con paraparesia4,10, ptosis palpebral sin oftalmoplejia4,9,10, saltatoria (afectación de miembros superiores y pares craneales exclusivamente)4,9, entre otras.
A menor edad del paciente pediátrico, mayores dificultades diagnósticas se plantean, sobre todo en aquellos casos en los que la sintomatología inicial se acerca a los límites que definen y delimitan esta patología. Así, por ejemplo, dolores musculares referidos, cojera, meningismo, cefalea, irritablilidad o somnolencia pueden ser síntomas que pueden aparecer en el inicio de estos pacientes. Ello obliga en muchas ocasiones a un amplio diagnóstico diferencial y a la realización de multitud de exploraciones complementarias antes de alcanzar el diagnóstico definitivo, lo que se podría evitar teniendo un alto índice de sospecha en este tipo de presentaciones atípicas y precoces de presentación del SGB3,4,14.
Los resultados del análisis del LCR (hiperproteinorraquia sin pleocitosis) y los estudios electrofisiológicos (disminución de la velocidad de conducción y de la amplitud del potencial evocado motor, latencia distal aumentada, bloqueo de la onda F o latencias mínimas de las ondas son hallazgos típicos que nos proporcionan el diagnóstico; sin embargo, hemos de tener en cuenta que estas características no se presentan en las fases iniciales de la enfermedad15.
A pesar de la complejidad clínica y el retraso diagnóstico que acontece en las edades más tempranas de presentación del SGB, la evolución habitual de estos niños es hacia la recuperación completa en semanas o meses.
ObjetivosVerificar la complejidad diagnóstica inicial y los distintos espectros clínicos en niños de edad menor de 6 años que presentaron SGB.
Pacientes y métodosEstudio retrospectivo y descriptivo de 8 pacientes diagnosticados de SGB a una edad inferior a 6 años en las últimas dos décadas en el Hospital General Universitario de Alicante e identificados en la base de datos hospitalaria con código IDC-9 357.0 (polineuropatía aguda infecciosa, postinfecciosa, SGB y síndrome de Miller Fisher). Incluimos a aquellos pacientes con criterios clínicos y paraclínicos de Asbury et al2,5,16 y neurofisiológicos descritos por Delense et al13,15. Se consideró con pronóstico favorable a aquellos con mejoría motora a los 15 días del diagnóstico y con buen pronóstico a aquellos con recuperación completa sin secuelas al año del diagnóstico.
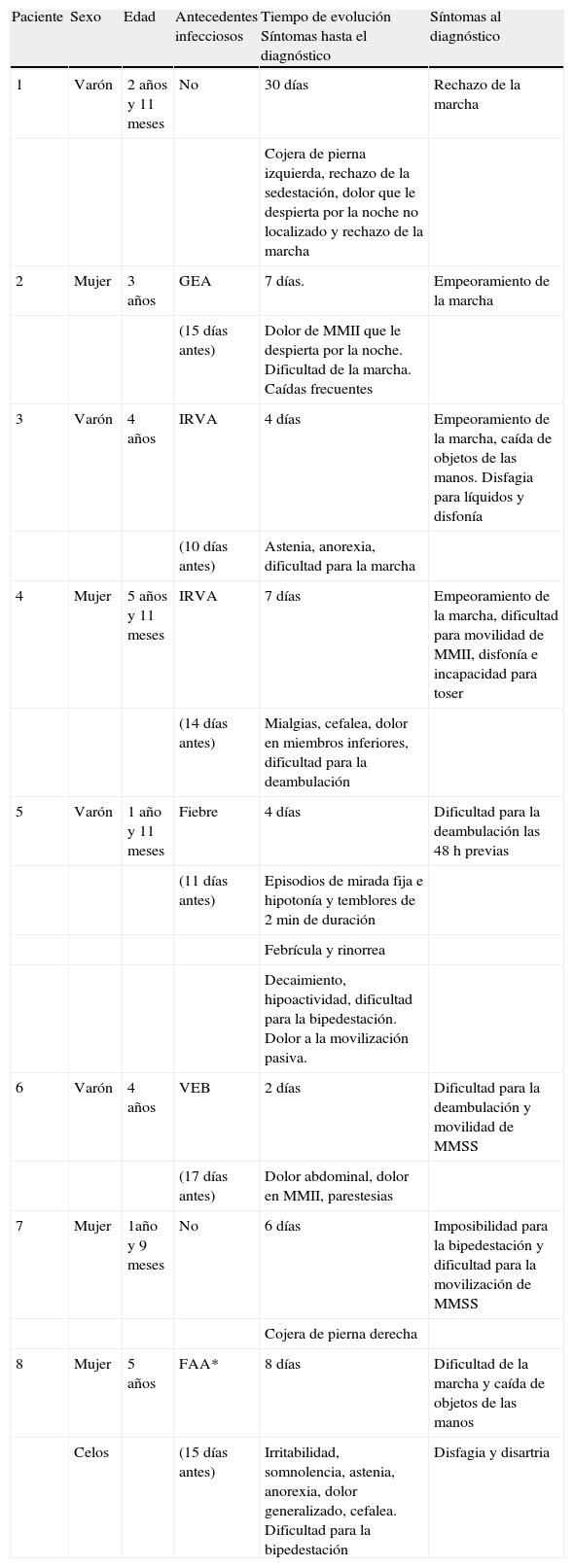
ResultadosSe incluyó a 8 pacientes (4 mujeres y 4 varones), con una edad media 3,5 años (intervalo entre 21 meses y 5 años y 11 meses). Las tablas 1 y 2 contienen lo más destacable de los resultados obtenidos. De cada paciente se valoró la edad de inicio de la sintomatología, la presencia de antecedentes personales infecciosos, el tiempo de evolución y los síntomas que precedieron al diagnóstico, la sintomatología en el momento del diagnóstico y la exploración neurológica objetivadas. Registramos las primeras hipótesis causales y las pruebas complementarias que se realizaron para descartarlas, así como los resultados del examen de LCR y estudios neurofisiológicos que determinaron el diagnóstico definitivo. Expondremos el tratamiento empleado y la evolución clínica.
SGB en menores de 6 años. Epidemiología, sintomatología inicial y al diagnóstico
| Paciente | Sexo | Edad | Antecedentes infecciosos | Tiempo de evolución Síntomas hasta el diagnóstico | Síntomas al diagnóstico |
| 1 | Varón | 2 años y 11 meses | No | 30 días | Rechazo de la marcha |
| Cojera de pierna izquierda, rechazo de la sedestación, dolor que le despierta por la noche no localizado y rechazo de la marcha | |||||
| 2 | Mujer | 3 años | GEA | 7 días. | Empeoramiento de la marcha |
| (15 días antes) | Dolor de MMII que le despierta por la noche. Dificultad de la marcha. Caídas frecuentes | ||||
| 3 | Varón | 4 años | IRVA | 4 días | Empeoramiento de la marcha, caída de objetos de las manos. Disfagia para líquidos y disfonía |
| (10 días antes) | Astenia, anorexia, dificultad para la marcha | ||||
| 4 | Mujer | 5 años y 11 meses | IRVA | 7 días | Empeoramiento de la marcha, dificultad para movilidad de MMII, disfonía e incapacidad para toser |
| (14 días antes) | Mialgias, cefalea, dolor en miembros inferiores, dificultad para la deambulación | ||||
| 5 | Varón | 1 año y 11 meses | Fiebre | 4 días | Dificultad para la deambulación las 48 h previas |
| (11 días antes) | Episodios de mirada fija e hipotonía y temblores de 2min de duración | ||||
| Febrícula y rinorrea | |||||
| Decaimiento, hipoactividad, dificultad para la bipedestación. Dolor a la movilización pasiva. | |||||
| 6 | Varón | 4 años | VEB | 2 días | Dificultad para la deambulación y movilidad de MMSS |
| (17 días antes) | Dolor abdominal, dolor en MMII, parestesias | ||||
| 7 | Mujer | 1año y 9 meses | No | 6 días | Imposibilidad para la bipedestación y dificultad para la movilización de MMSS |
| Cojera de pierna derecha | |||||
| 8 | Mujer | 5 años | FAA* | 8 días | Dificultad de la marcha y caída de objetos de las manos |
| Celos | (15 días antes) | Irritabilidad, somnolencia, astenia, anorexia, dolor generalizado, cefalea. Dificultad para la bipedestación | Disfagia y disartria |
FAA: faringoamigdalitis aguda; GEA: gastroenteritis aguda; IRSV: infección de vías respiratorias superiores; MMII: miembros inferiores; MMSS: miembros superiores; VEB: infección por virus de Epstein-Barr.
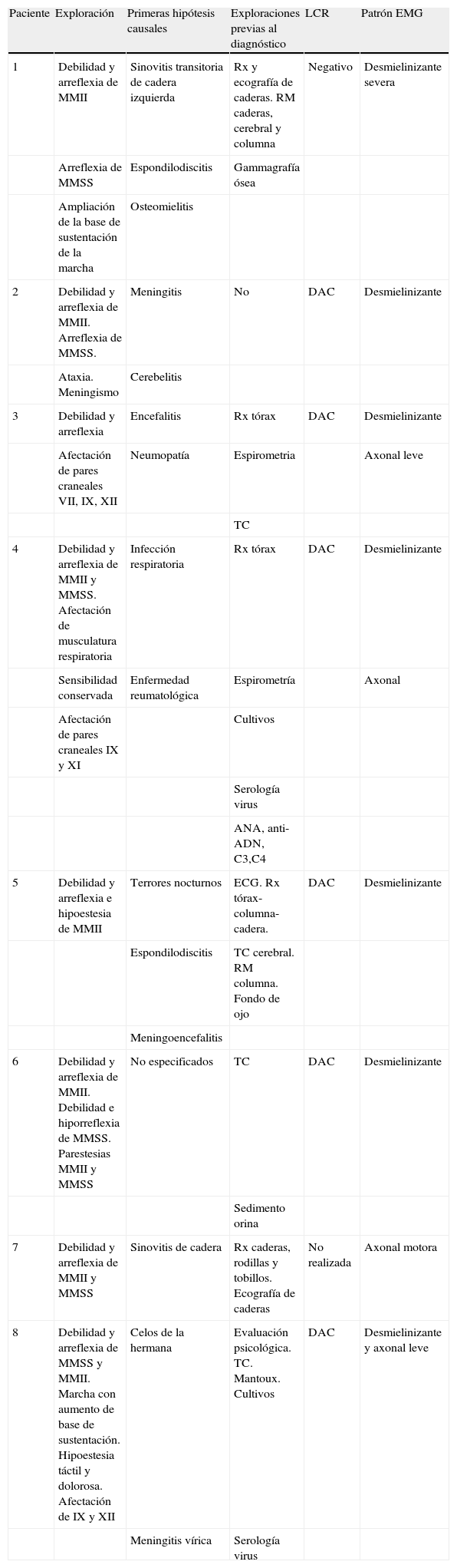
SGB en menores de 6 años. Exploración, diagnóstico diferencial, exploraciones complementarias
| Paciente | Exploración | Primeras hipótesis causales | Exploraciones previas al diagnóstico | LCR | Patrón EMG |
| 1 | Debilidad y arreflexia de MMII | Sinovitis transitoria de cadera izquierda | Rx y ecografía de caderas. RM caderas, cerebral y columna | Negativo | Desmielinizante severa |
| Arreflexia de MMSS | Espondilodiscitis | Gammagrafía ósea | |||
| Ampliación de la base de sustentación de la marcha | Osteomielitis | ||||
| 2 | Debilidad y arreflexia de MMII. Arreflexia de MMSS. | Meningitis | No | DAC | Desmielinizante |
| Ataxia. Meningismo | Cerebelitis | ||||
| 3 | Debilidad y arreflexia | Encefalitis | Rx tórax | DAC | Desmielinizante |
| Afectación de pares craneales VII, IX, XII | Neumopatía | Espirometria | Axonal leve | ||
| TC | |||||
| 4 | Debilidad y arreflexia de MMII y MMSS. Afectación de musculatura respiratoria | Infección respiratoria | Rx tórax | DAC | Desmielinizante |
| Sensibilidad conservada | Enfermedad reumatológica | Espirometría | Axonal | ||
| Afectación de pares craneales IX y XI | Cultivos | ||||
| Serología virus | |||||
| ANA, anti-ADN, C3,C4 | |||||
| 5 | Debilidad y arreflexia e hipoestesia de MMII | Terrores nocturnos | ECG. Rx tórax- columna-cadera. | DAC | Desmielinizante |
| Espondilodiscitis | TC cerebral. RM columna. Fondo de ojo | ||||
| Meningoencefalitis | |||||
| 6 | Debilidad y arreflexia de MMII. Debilidad e hiporreflexia de MMSS. Parestesias MMII y MMSS | No especificados | TC | DAC | Desmielinizante |
| Sedimento orina | |||||
| 7 | Debilidad y arreflexia de MMII y MMSS | Sinovitis de cadera | Rx caderas, rodillas y tobillos. Ecografía de caderas | No realizada | Axonal motora |
| 8 | Debilidad y arreflexia de MMSS y MMII. Marcha con aumento de base de sustentación. Hipoestesia táctil y dolorosa. Afectación de IX y XII | Celos de la hermana | Evaluación psicológica. TC. Mantoux. Cultivos | DAC | Desmielinizante y axonal leve |
| Meningitis vírica | Serología virus |
ANA: anticuerpos antinucleares; anti-ADN: anticuerpos anti-ADN; DAC: disociación albumino-citológica; MMII: miembros inferiores; MMSS: miembros superiores; RM: resonancia magnética; Rx: radiología; TC: tomografía computerizada.
Ningún paciente presentó antecedentes familiares neurológicos de interés.
En 6 pacientes se registró la presencia de una infección previa a las manifestaciones clínicas sugestivas de SGB (75%). Se registran gastroenteritis aguda, infección de vías respiratorias altas, mononucleosis por virus de Epstein-Barr, fiebre sin foco y faringoamigdalitis. El tiempo medio de estas infecciones previo a los síntomas neurológicos fue de 16,5 días (tabla 1).
Cuadro clínico y tiempo de evolución al diagnósticoLa sintomatología que precedió al diagnóstico fue de carácter muy heterogéneo. Inicialmente, encontramos sintomatología inespecífica como irritabilidad, somnolencia, astenia, anorexia, cojera, dolor no localizado, cefalea y rechazo de la marcha.
El tiempo medio de evolución de las manifestaciones clínicas fue de 8,5 días. En el momento del diagnóstico en todos los casos se describe el máximo de afectación clínica excepto en uno de los casos. Se trata de la paciente de mayor edad incluida en el estudio, en la que en el momento del ingreso se describe disminución de la fuerza en miembros inferiores con dificultad progresiva para la deambulación con caídas al suelo de 8 días de evolución, con empeoramiento en las últimas 24h tanto de la marcha como por la aparición de debilidad ascendente de miembros superiores, disfonía, disfagia y dolor, sin parestesias. A su ingreso en la unidad de cuidados intensivos pediátrica (UCIP), continúa la progresión afectándose la musculatura respiratoria, precisando ventilación mecánica durante 4 días. Tras el tratamiento con dosis altas de inmunoglobulinas, mejora rápidamente, siendo dada de alta de la UCIP a los 7 días, con mejora progresiva en la planta de escolares con hiporreflexia exclusivamente a los 15 días.
Los síntomas que abocaron al diagnóstico fueron mayoritariamente empeoramiento en la dificultad para la marcha y, en algunos casos, dificultad para la movilización de miembros superiores o para la sujeción de objetos (75%). En dos pacientes se objetivó disfagia y disfonía (tabla 1).
Examen neurológico (tabla 2)Manifestaciones motorasTodos los pacientes incluidos en el estudio presentaron debilidad o paresia con arreflexia de miembros inferiores. Uno de ellos tuvo un inicio asimétrico (hemiparesia izquierda) durante los primeros 10 días, progresando hacia la simetría; 6 de los pacientes presentaron debilidad o paresia de miembros superiores (75%). Sólo un paciente precisó ventilación mecánica por afectación de la musculatura respiratoria (12%).
Manifestaciones sensitivasSe registraron síntomas sensitivos en 5 historias clínicas (62,5%). En 3 pacientes se encontraron parestesias, 2 de ellas asociadas a dolor (37,5%) y 5 de los pacientes refirieron dolor sin localización determinada (62,5%).
Afectación de pares cranealesSe encontró en 3 pacientes (37,5%). Los pares afectados fueron VII, IX, XI y XII.
Otras manifestacionesSe describió ataxia como aumento de la base de sustentación e inestabilidad de la marcha en uno de los pacientes (12%). En un paciente se objetivó temblor (12%).
Se encontraron signos meníngeos positivos en 2 pacientes (25%).
No se registraron manifestaciones autonómicas, fiebre, vómitos, diarrea o depresión neuropsíquica en ningún caso.
Diagnóstico diferencialDada la complejidad de estos pacientes y la sintomatología tan inespecífica inicial, las primeras hipótesis causales que se describen en estos pacientes previo al diagnóstico de SGB fueron: problemas psicológicos (terrores nocturnos, celos), enfermedades infecciosas del sistema nervioso central (meningitis, meningoencefalitis, cerebelitis, espondilodiscitis), enfermedades reumatoideas (artritis reumatoide juvenil y sinovitis de cadera.
Pruebas complementarias (tabla 2)Primeras exploraciones complementariasPara descartar las primeras hipótesis causales se realizaron múltiples pruebas complementarias en 7 de los pacientes. Entre estas pruebas encontramos: radiología simple (tórax, caderas, columna), ecografía de caderas, tomografía computarizada craneal, resonancia magnética (cerebral, columna, caderas), gammagrafía ósea, electrocardiograma y espirometría, cultivos y serología, pruebas de cribado reumatológico.
Estudios de líquido cefalorraquídeoEn 7 de los casos se registraron resultados del estudio citoquímico del LCR. En 6 de los casos se realizó después de la primera semana de evolución, encontrándose disociación albuminocitológica en 5 de ellos (83,3%). De éstos, fue negativa en el paciente diagnosticado más tardíamente, habiéndose realizado a los 17 días de progresión del cuadro. Sólo en uno de los pacientes se realizó el estudio de LCR antes de la primera semana de evolución, a los 4 días; en este caso, también se objetivó hiperproteinorraquia con normalidad celular.
Estudios electrofisiológicosEn el 100% de los pacientes se realizó un estudio de velocidad de conducción y electromiograma, siendo positivos en todos los casos. El patrón más frecuente informado fue el desmielinizante con distintos grados de gravedad, en 7 de los pacientes (87,5%). En 4 de ellos se registró un patrón desmielinizante puro (50%), mientras que en 3 pacientes se añadió un componente axonal leve (38%). Sólo uno de los casos se describe un patrón axonal motor puro (12%).
A ningún paciente se le ha realizado la determinación de anticuerpos antifosfolípidos.
TratamientoSiete de los pacientes fueron tratados con inmunoglobulinas. Ninguno fue tratado con plasmaféresis. Un paciente no recibió tratamiento debido a que su diagnóstico fue a las dos semanas de evolución, en fase de mejoría clínica con deambulación. Solo una paciente precisó ventilación mecánica.
En dos de los pacientes se inició tratamiento antibiótico intravenoso al ingreso, previo al diagnóstico definitivo (tabla 3).
SGB en menores de 6 años. Tratamiento y evolución
| Pacientes | Tratamiento | Evolución |
| 1 | No | Máxima afectación: 7días |
| Revisiones | ||
| 15 días: hiporreflexia y leve debilidad de MMII | ||
| Mes: hiporreflexia | ||
| 6 meses y año: asintomático | ||
| 2 | IG 5 días | Máxima afectación: 8 días |
| Revisiones: | ||
| 15 días: hiporreflexia y debilidad | ||
| Mes: hiporreflexia | ||
| 6 meses y año: asintomático | ||
| 3 | IG 5 días | Máxima afectación: 6 días |
| Revisiones | ||
| 15 días: parálisis facial en recuperación e hiporreflexia | ||
| Mes: hiporreflexia y mínima afectación facial | ||
| 6 meses: hiporreflexia | ||
| Año: asintomático | ||
| 4 | Antibioterapia VM 4días | Máxima afectación: 8 días |
| IG 5 días | Revisiones | |
| 15 días y mes: hiporreflexia y debilidad | ||
| 6 mes y año: asintomática | ||
| 5 | Antibioterapia | Máxima afectación: 5 días |
| IG 5 días | Revisiones | |
| 15 días: arreflexia, hipoestesia y molestias en MMII | ||
| Mes y 6 meses: hiporreflexia | ||
| Año: asintomático | ||
| 6 | IG 5 días | Máxima afectación: 4 días |
| Revisiones | ||
| 15 días: leves parestesias y dolor. Debilidad de y parestesias en MMII. Mínimo temblor | ||
| Mes: hiporreflexia y leve debilidad | ||
| 6 meses: hiporreflexia | ||
| Año: asintomático | ||
| 7 | IG 5 días | Máxima afectación: 7 días |
| Revisiones | ||
| 15 días y mes: hiporreflexia | ||
| 6 meses y año: asintomático | ||
| 8 | IG 5 días | Máxima afectación: 10 días |
| Revisiones | ||
| 15 días y mes: hiporreflexia | ||
| 6 meses y año: asintomática |
IG: inmunoglobulinas; VM: ventilación mecánica; MMII: miembros inferiores.
La máxima afectación clínica se encuentra a los 8 días de media. A los 15 días de evolución, la mejoría en todos los pacientes es significativa, quedando como sintomatología residual hiporreflexia vs hiporreflexia y debilidad de miembros inferiores (menor que al diagnóstico). Al mes de evolución, la sintomatología más frecuente fue hiporreflexia, añadiéndose mínima debilidad de miembros inferiores en cuatro de los pacientes y parálisis facial en recuperación en uno de los casos. A los 6 meses del diagnóstico, sólo en un paciente persistía una mínima hiporreflexia, siendo al año cuando todos los pacientes se encuentran totalmente asintomáticos (tabla 3).
DiscusiónEl diagnóstico precoz de la causa más frecuente de parálisis flácida en la infancia suele ser difícil y requerir un alto índice de sospecha clínica. Los 8 pacientes incluidos en este estudio así lo corroboran, especialmente si atendemos al tiempo de evolución de la sintomatología variada con la que comienzan hasta el momento del diagnóstico y la multitud de exploraciones complementarias que se les practican hasta confirmar el SGB. La forma de presentación que prevalece en las edades de mayor incidencia (parálisis flácida arrefléctica de inicio agudo y progresiva, ascendente, acompañada de parestesias y afectación del sistema nervioso vegetativo) pueden no ser la forma habitual con que se manifiesta en pacientes preescolares o de edades incluso inferiores.
En cuanto los antecedentes, en nuestro estudio se confirma la asociación a un proceso infeccioso previo en dos tercios de los pacientes como se describe en otras publicaciones1–4.
Hay que tener en cuenta que los primeros síntomas podrán ser muy variados e inespecíficos, como puede ser dolor, meningismo (30%)4, cefalea, irritabilidad, somnolencia o ataxia, que nos hacen plantearnos al inicio de cuadro distintos diagnósticos diferenciales como encefalopatías agudas8, o distintas causas de ataxia13. Otros diagnósticos diferenciales a tener en cuenta son neuropatías periféricas, alteraciones de la unión neuromuscular, enfermedades musculares y mielopatías agudas como la compresión medular4,16. Esto implica una situación de falta de sospecha clínica y un retraso en el tratamiento.
En nuestro trabajo se confirma la gran heterogenicidad de las manifestaciones iniciales, lo que implica un diagnóstico diferencial muy amplio previo al diagnóstico y la realización de múltiples pruebas complementarias innecesarias que aumentan la iatrogenia y el gasto sanitario. La orientación diagnóstica correcta generalmente aparece al empeorar la sintomatología de los pacientes, sobre todo a nivel de empeoramiento de la marcha. A la exploración destaca la afectación motora de miembros inferiores de todos los pacientes con arreflexia, a lo que se añade hipoestesia, afectación de pares craneales, ataxia y/o meningismo en algunos casos.
Los criterios diagnósticos se centran en los hallazgos cliniconeurofisiológicos y del LCR, aunque últimamente se añade al diagnóstico la determinación de anticuerpos antigangliósido que se han encontrado asociados a las variantes menos frecuentes del SGB. Estos anticuerpos podrían estar relacionados con una determinada evolución clínica. El patrón electrofisiológico de polineuropatía sensitivo-motora desmielinizante se confirma como el patrón clásico habitual en edades precoces y con un excelente pronóstico a corto y medio plazo.
La evolución clínica de la globalidad del grupo es muy satisfactoria prácticamente desde el segundo mes de inicio de la sintomatología variada descrita, quedando la arreflexia y la actividad motora (correr principalmente) normalizada a los 6 meses de evolución, aproximadamente. Al año, la normalidad es completa en todos los pacientes.
Se ha descrito que en la infancia el pronóstico del SGB es presumiblemente mejor que en adultos, siendo la mortalidad de entre 1-5% y con un porcentaje de secuelas, generalmente leves, de un 25%, y con recuperación motora relativamente más rápida17–19. Se ha descrito que la variante axonal es la variante más grave, con tiempo de recuperación motora mayor13, mientras en otras publicaciones consideran que esta variante tiene un pronóstico similar a la desmielinizante13. También se ha intentado relacionar la gravedad del déficit motor y el tiempo de meseta con el pronóstico funcional de los niños20,21. En nuestro estudio, se confirma el excelente pronóstico en todos los pacientes (incluido la variante axonal pura).
En resumen, los 8 pacientes descritos en nuestro estudio nos indican que en todo paciente de edad inferior a la escolar con un cuadro agudo de sintomatología en miembros inferiores, tanto motora como sensitiva, que asocie arreflexia osteotendinosa debería sospecharse precozmente esta patología subsidiaria del tratamiento con inmunoglobulinas por vía intravenosa, aunque inicialmente los estudios de LCR y electrofisiológicos no sean diagnósticos, con el objetivo de evitar la posible progresión clínica que podría requerir cuidados intensivos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
