



Introducción
El síndrome de Crigler-Najjar (SCN) es un trastorno del metabolismo de una bilirrubina consistente en un déficit de la enzima UDP-glucuroniltransferasa (UGT) de forma total (tipo 1) o parcial (tipo 2) 1. De las isoformas de dicha enzima, sólo la UGT1A1 es la que contribuye de forma significativa al proceso de conjugación de la bilirrubina en humanos. En el SCN tipo 1 el defecto consiste en deleciones, mutaciones o inserciones en cualquiera de los 5 exones que constituyen el gen UGT1A1 del brazo largo del cromosoma 2, con lo que se producirán codones stop prematuros o sustituciones de un aminoácido que impedirán la adecuada transcripción del ARNm y producirán la enfermedad mediante su transmisión autosómica recesiva 2,3. Por su parte, en el SCN tipo 2 la alteración consiste en la sustitución de un aminoácido, con lo que se reduce la actividad catalítica de la enzima, mientras que en el síndrome de Gilbert lo que ocurre es que el gen UGT1A1 contiene una secuencia TATAA más 4,5.
El SCN fue descrito por primera vez en 1952 por Crigler y Najjar 6, al referirse a 6 niños de 3 familias que presentaban durante los primeros días de vida ictericia grave con predominio de la bilirrubina indirecta y que fallecían por querníctero antes de los 2 años de edad. Se considera que el SCN tipo 1 tiene una frecuencia estimada en 0,6 casos por millón de habitantes, y hasta el momento se han recogido en la literatura médica unos 70 casos 7.
Si el déficit de la enzima es total (SCN tipo 1) la concentración de bilirrubina indirecta (BI) supera con frecuencia los 20 mg/dl, no existen restos de bilirrubina conjugada en el aspirado duodenal y no hay respuesta al tratamiento con enzimas inductoras de la UGT como el fenobarbital 8,9. En estos pacientes el riesgo de querníctero es importante, lo que ensombrece su pronóstico si no se instaura un tratamiento 6,10. Esta complicación neurológica constituye el principal riesgo en el tratamiento de estos niños, de modo que se produce una necrosis celular en los ganglios de la base, el hipocampo, el núcleo cerebeloso y la sustancia gris 6,10,11, producida por el depósito de BI que, cruzando a altas dosis la barrera hematoencefálica, inhibe la síntesis de ARN y proteínas, y altera el metabolismo de los hidratos de carbono y la respiración mitocondrial. Por el contrario, cuando el déficit de la enzima es parcial (SCN tipo 2) la BI rara vez supera los 20 mg/dl, existe respuesta al fenobarbital y el curso es más benigno y con pocas probabilidades de desarrollar querníctero 12 (tabla 1).

El tratamiento consiste en la aplicación de fototerapia 13, que convierte la fracción IXalfa-ZZ de la BI en un isómero capaz de ser excretado por la bilis. Pueden asociarse quelantes biliares como las sales cálcicas o la colestiramina, y, ante una elevación aguda de la BI que pudiera producir alteraciones neurológicas irreversibles, puede emplearse la plasmaféresis para reducir rápidamente las concentraciones de BI. No obstante, el único tratamiento curativo existente en la actualidad es el trasplante hepático 14,15. El objetivo del estudio es analizar la evolución de los pacientes diagnosticados de SCN.
Pacientes y métodos
Se revisan las historias clínicas de 7 pacientes diagnosticados de SCN tipo 1 que han sido seguidos por el servicio de hepatología de nuestro hospital entre los años 1987 y 2004. Se recogen: la fecha de nacimiento y el sexo, la existencia de antecedentes familiares de consanguinidad o de otras enfermedades de transmisión genética, la edad de diagnóstico, el tiempo de seguimiento, el método empleado para hacer el diagnóstico y el curso clínico, haciendo énfasis en la situación neurológica y en la variación de las cifras de BI en función de las enfermedades intercurrentes y de los tratamientos pautados.
Resultados
Los pacientes fueron 3 niños y 4 niñas, dos de ellas gemelas homozigotas. En dos familias existía consanguinidad en primer y cuarto grado y en 4 casos había hermanos sanos; el resto eran hijos únicos. De los niños, tres tenían algún familiar diagnosticado de enfermedad de Gilbert: en un caso la madre, en otro la madre y una prima materna y en un tercero un primo lejano paterno. Del mismo modo, en las pacientes gemelas homozigotas tanto los padres como los 2 hermanos sanos tenían cifras de BI superiores a 1,5 mg/dl en repetidas ocasiones.
La ictericia fue observada en todos los niños durante los primeros 3 días de vida, y el ingreso que motivó la sospecha diagnóstica de SCN se produjo entre el 4.º y el 60.º día (media de 19,5 días). En ese momento los pacientes tenían unas cifras de BI de entre 12,5 y 32 mg/dl (media de 20 mg/dl). En todos ellos los parámetros de función hepática fueron normales y no existieron signos de hemólisis.
La confirmación diagnóstica se realizó en cuatro de los casos mediante el estudio de las alteraciones del gen UGT1A1 (tabla 2). En uno de los pacientes se detectó una alteración de la inserción para 4 nucleótidos (TACC) después del nucleótido 490 en el exón 1 del gen, y el paciente era homozigoto para dicho defecto y cada uno de los padres heterozigoto. Otro de los pacientes resultó ser heterozigoto compuesto para las mutaciones nt609del24bp (procedente de la madre) y 1305-2 A > G (procedente del padre), pero sin haber sido descritas ninguna de estas mutaciones hasta entonces. Por último, las dos gemelas resultaron ser homozigotas para la mutación R336W del mismo gen. De los 3 casos restantes, uno se diagnóstico al determinar la ausencia total de actividad de la enzima UGT en tejido hepático, mientras que los otros 2 niños fueron diagnosticados ambos de SCN ante la inexistencia de bilirrubina conjugada en el aspirado duodenal y ante la falta de respuesta al tratamiento con fenobarbital.

El seguimiento medio de los pacientes en el servicio fue de 8,3 años (oscilando desde los 14 meses a los 17 años). La edad actual de los niños está comprendida desde los 2 años a los 20 años y 8 meses (media de 10,9 años).
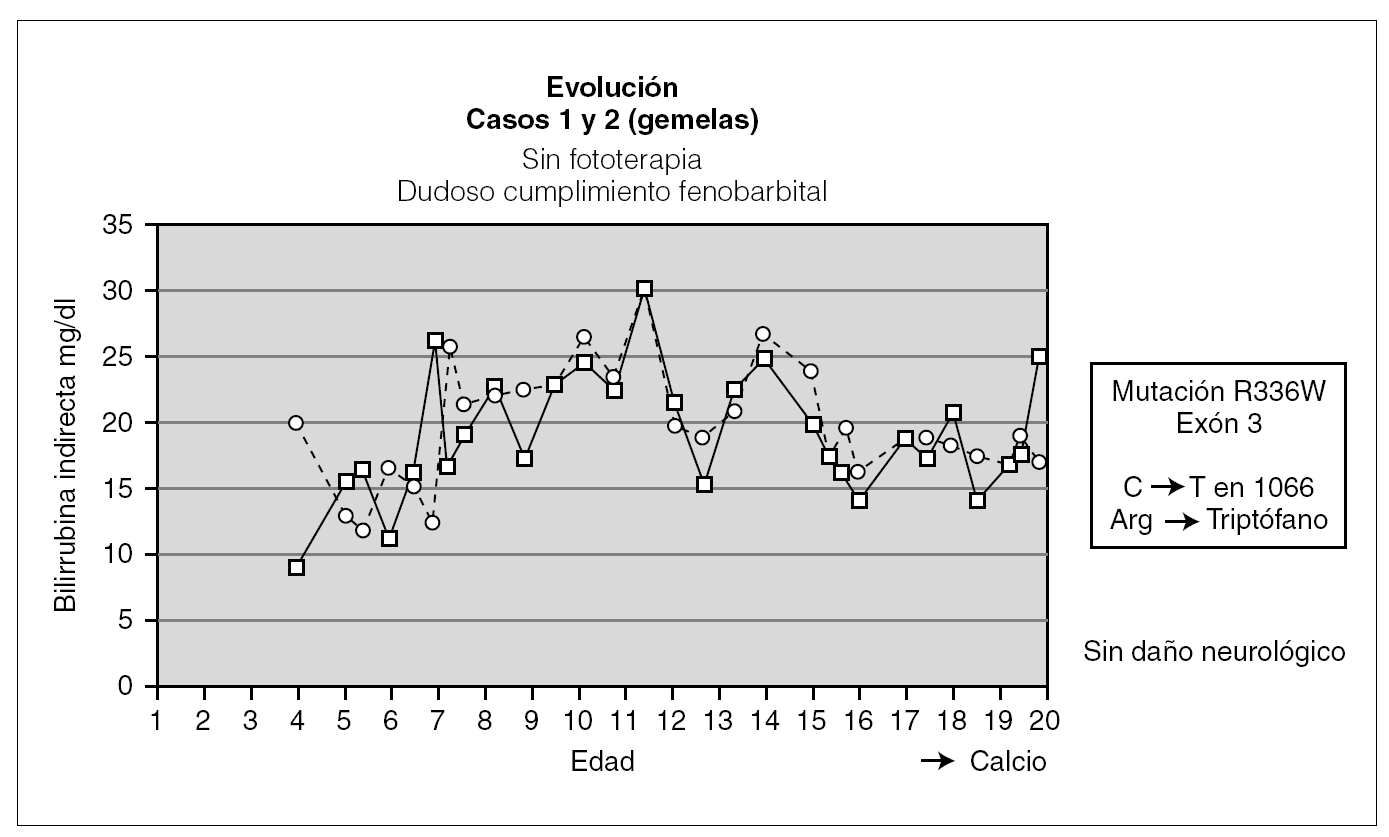
En las 2 pacientes gemelas homozigotas el seguimiento ha sido hasta los 20 años de edad. Fueron diagnosticadas al final del primer mes de vida, y han sido tratadas desde entonces con fenobarbital con respuesta parcial al mismo. Durante el seguimiento han tenido fluctuaciones de la BI en función del cumplimiento del tratamiento y de la aparición de procesos intercurrentes (fig. 1). Una de las pacientes fue diagnosticada a los 13 años de una púrpura trombocitopénica idiopática resistente al tratamiento con corticoides y con gammaglobulinas, y que precisó para su curación la esplenectomía; otra paciente presenta litiasis biliar. Hasta el momento no se ha planteado en estas pacientes el trasplante hepático al tener un desarrollo psicomotor normal con cifras de BI mantenidas durante la mayor parte de su evolución por debajo de 25 mg/dl.
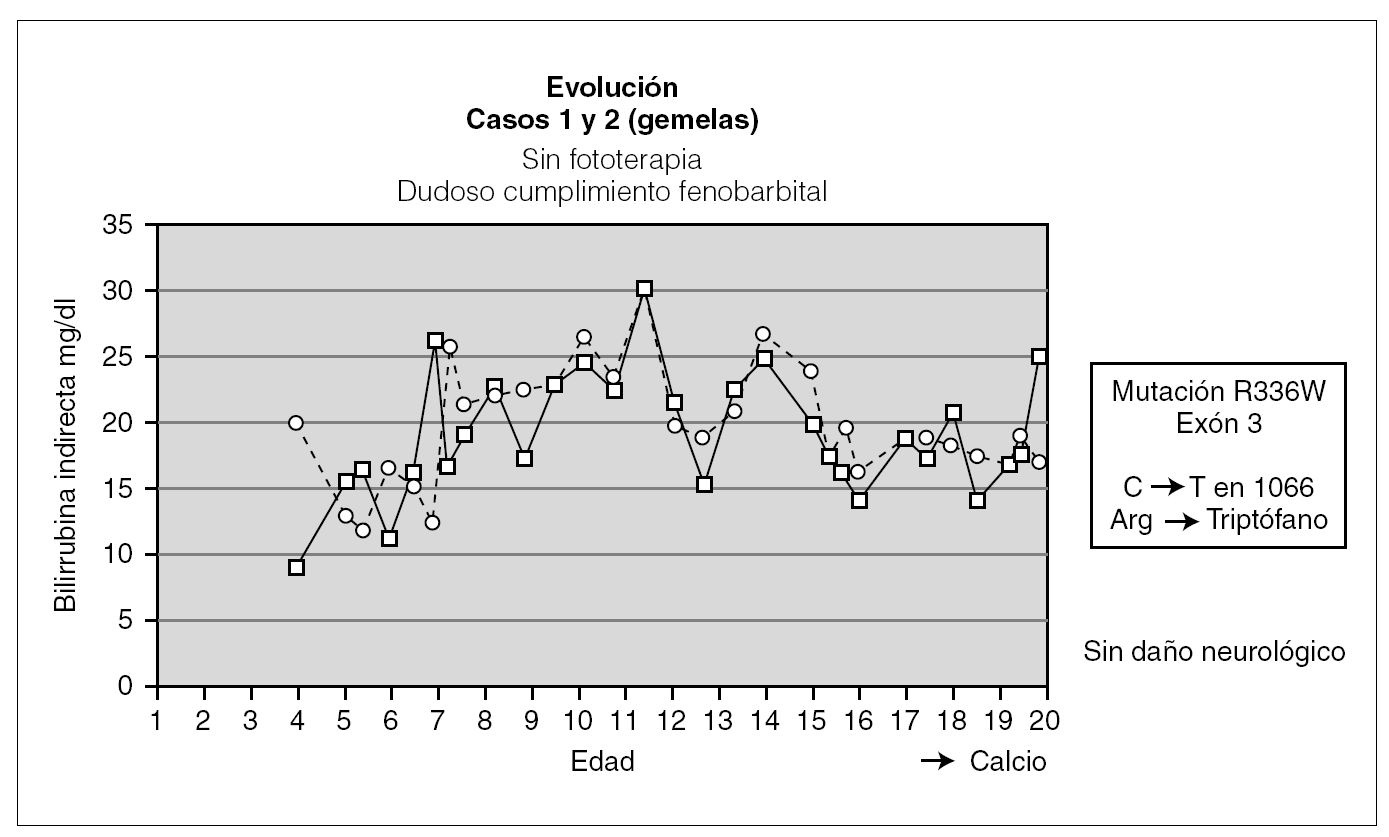
Figura 1. Evolución de las dos gemelas.
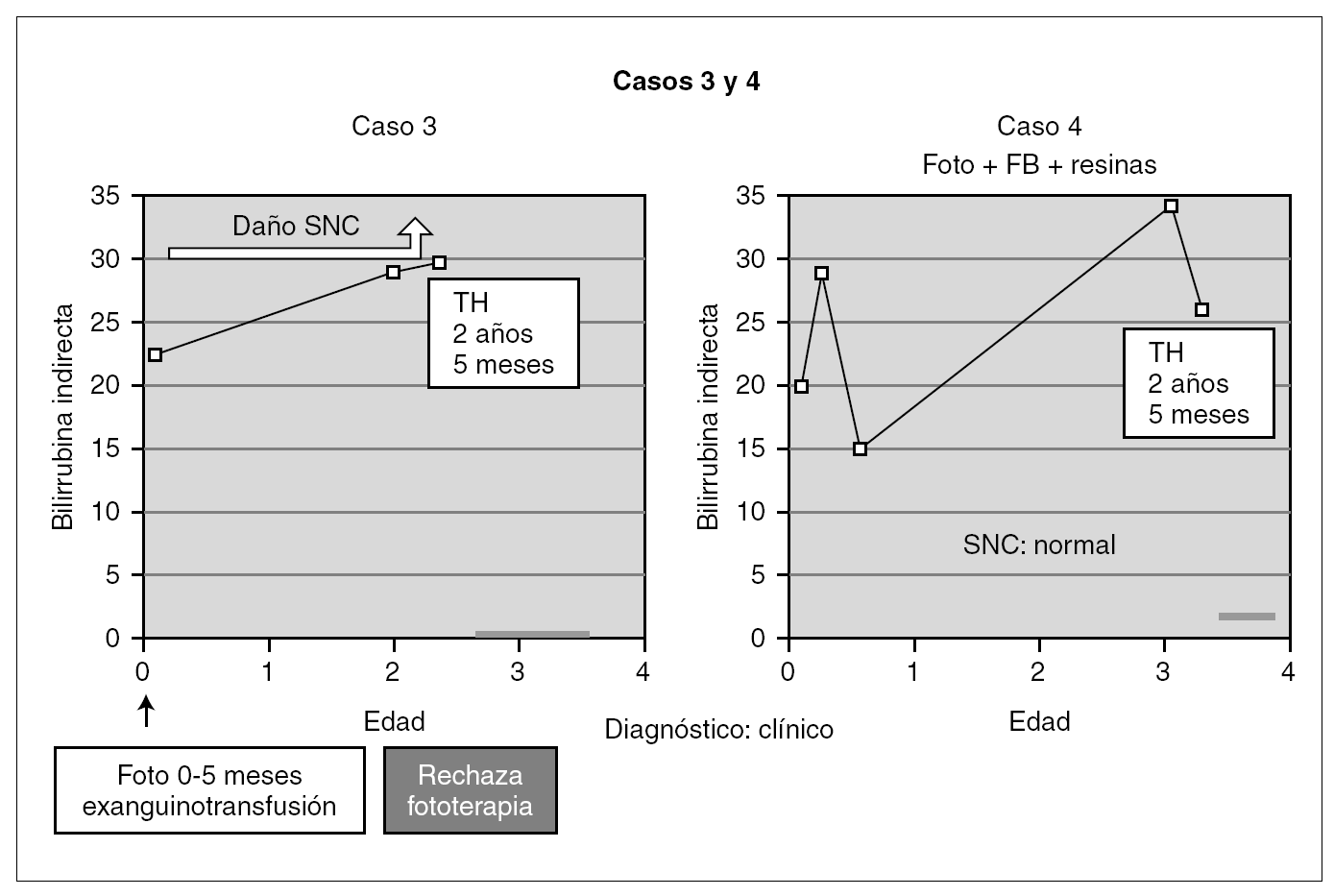
Entre los 2 pacientes trasplantados (fig. 2), el diagnóstico también se realizó en el primer mes de vida, y se inició tratamiento con fototerapia y fenobarbital. En uno de ellos el trasplante fue realizado a los 2 años y 5 meses, tras evidenciarse BI persistentemente elevada por encima de 25 mg/dl con rechazo de la fototerapia por parte de los padres y signos de deterioro neurológico progresivo con hipotonía axial, incapacidad para la sedestación y deambulación, pérdida del lenguaje y crisis convulsivas. En el electroencefalograma (EEG) se apreció actividad cerebral retardada generalizada con lesiones visibles en resonancia magnética (RM) en la sustancia blanca bifrontal, biparietal, así como en el tálamo y y el caudado (fig. 3). Del mismo modo, se detectó un retardo importante en la respuesta a los estímulos de las vías auditivas y visuales. A pesar del buen control de la BI tras el trasplante, la paciente continuó con un retraso psicomotor grave y falleció a los 9 meses de la intervención debido a un síndrome de Budd-Chiari en el injerto. El otro se trasplantó a los 3 años y 4 meses de edad ante el mal control de las cifras de BI con fototerapia. Con posterioridad al trasplante se normalizó el metabolismo de la bilirrubina y tuvo una evolución favorable.
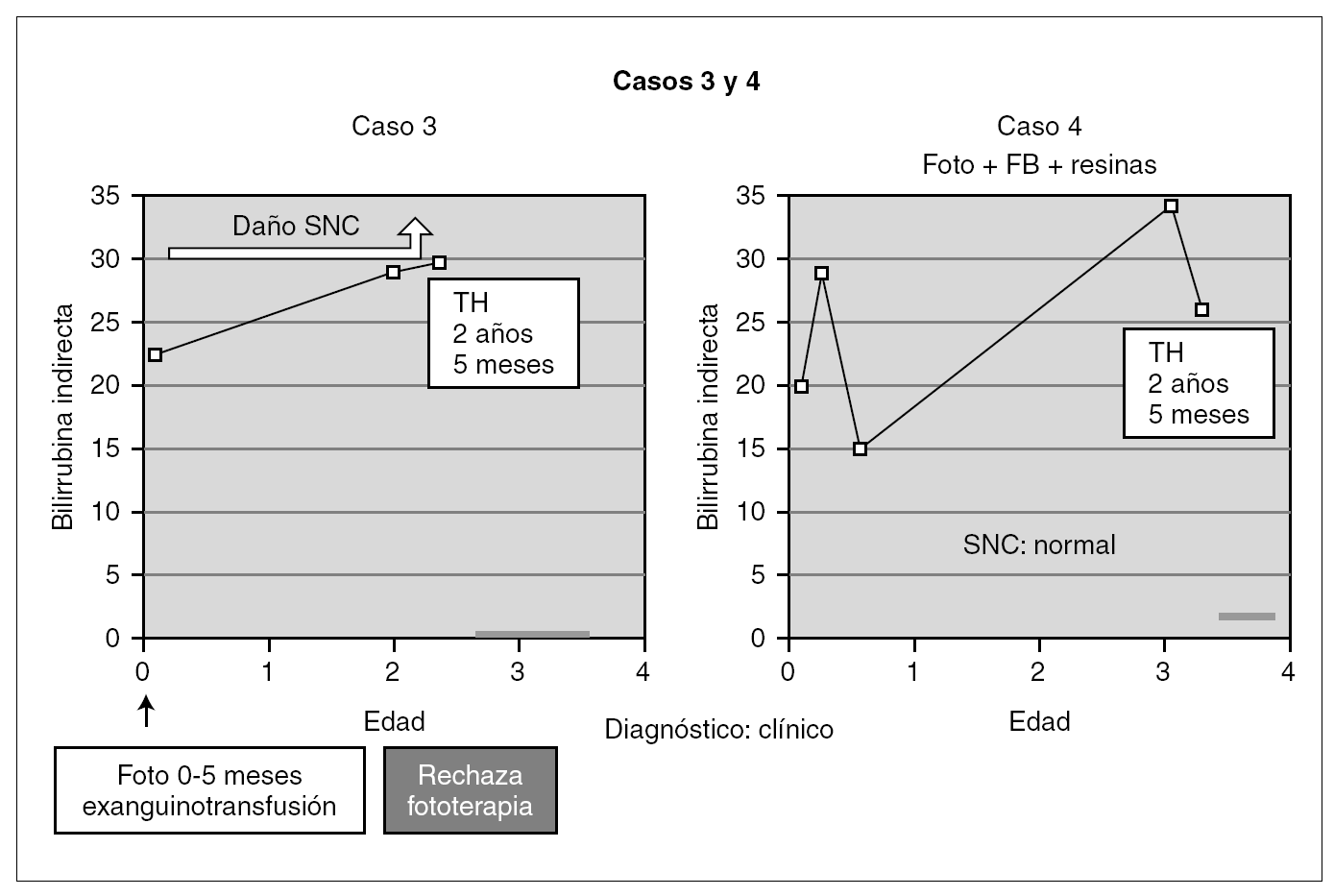
Figura 2. Evolución de los 2 pacientes trasplantados previa a la intervención (foto: fototerapia).
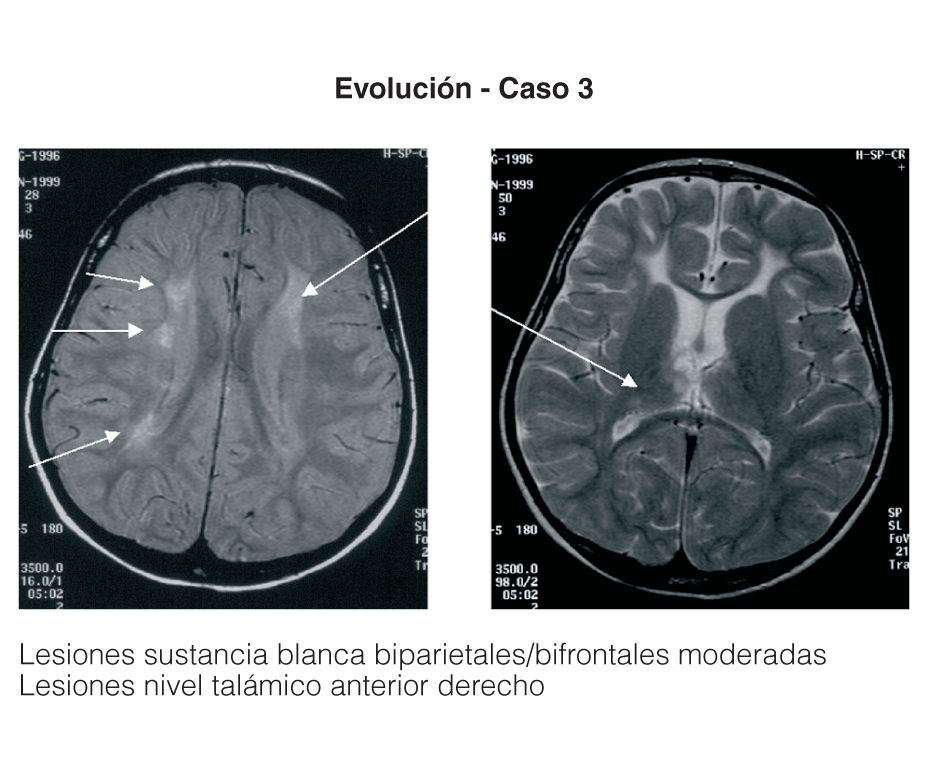
Figura 3. Resonancia magnética de la paciente con querníctero trasplantada.
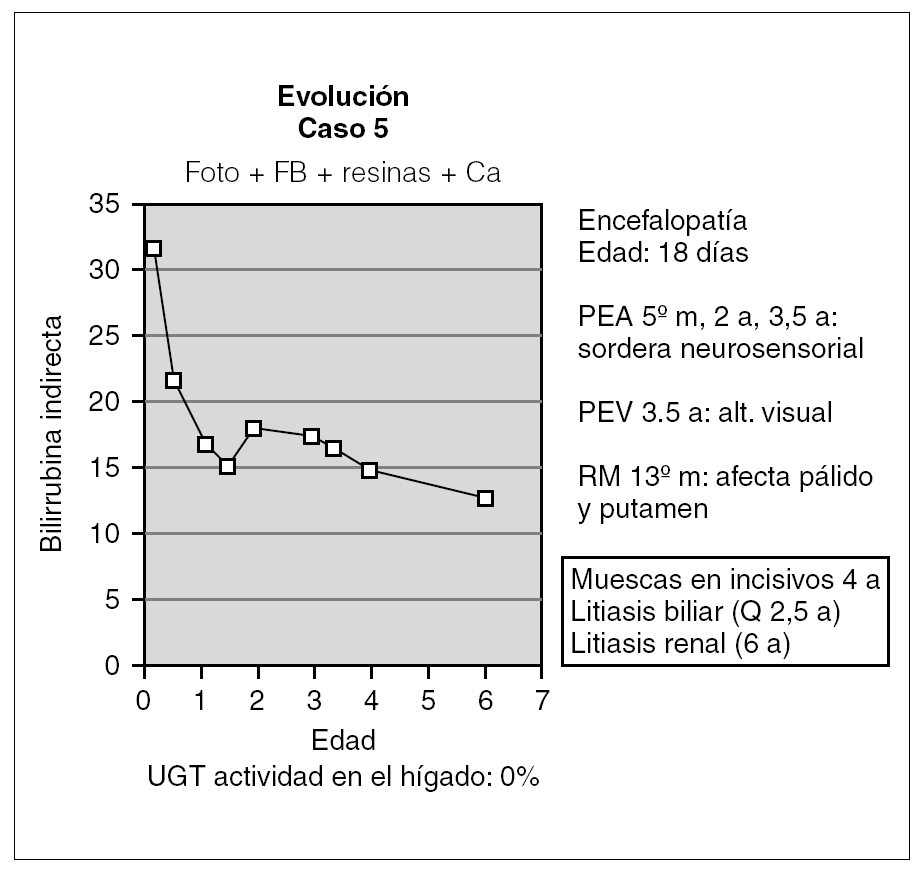
Otro de los pacientes se diagnosticó a los 18 días de vida por la aparición de alteraciones neurológicas coincidiendo con un pico de BI de 32 mg/dl (fig. 4). A partir de ahí comenzó con irritabilidad intensa y crisis convulsivas, para continuar con hipotonía marcada y retraso psicomotor grave. En el EEG se detectó déficit de organización e integración de la actividad eléctrica cerebral, en la RM se observó aumento de señal en los ganglios basales (putamen y pálido) por gliosis sin hallarse respuesta en los potenciales evocados de la vía auditiva y visual. Desde entonces, se ha mantenido tratamiento con fototerapia entre 6 y 10 h diarias y con quelantes de sales biliares, por lo que se mantiene con BI < 25 mg/dl. Así mismo, durante la evolución ha desarrollado litiasis biliar y renal asintomáticas y se ha realizado una colecistectomía a los 2 años y 6 meses. Posteriormente, a partir de los 4 años de edad, se han evidenciado muescas en los incisivos superiores. En cuanto a la evolución neurológica, no se ha producido recuperación alguna de su retraso psicomotor.
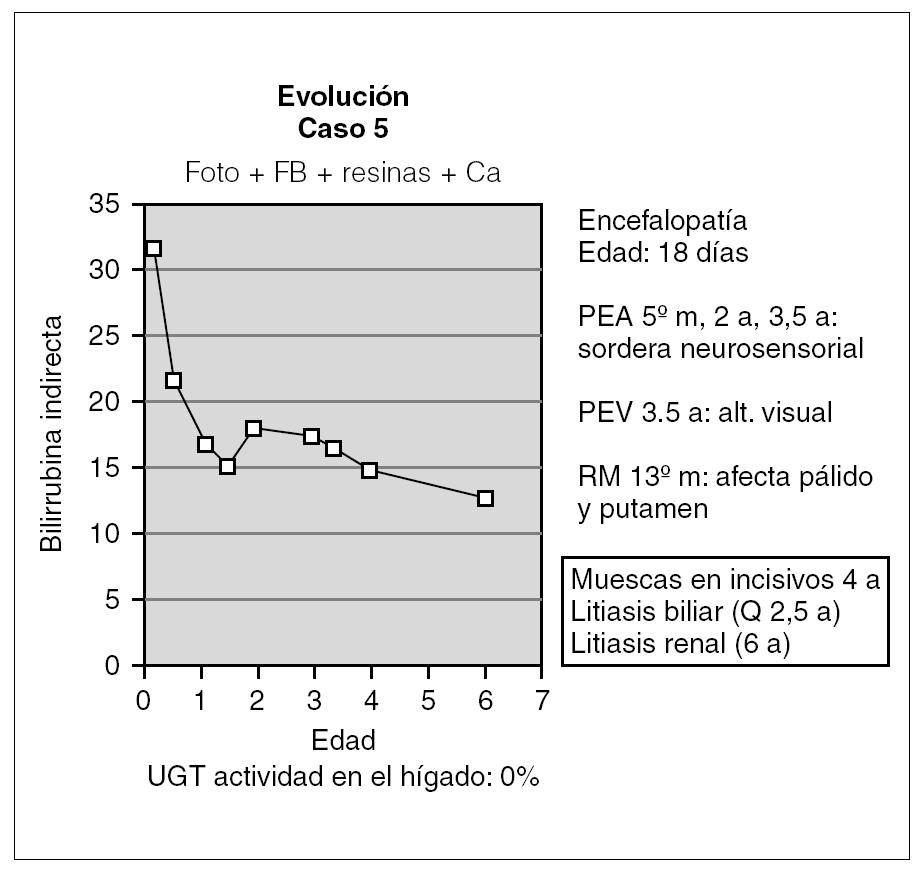
Figura 4. Evolución del paciente que presentó querníctero.
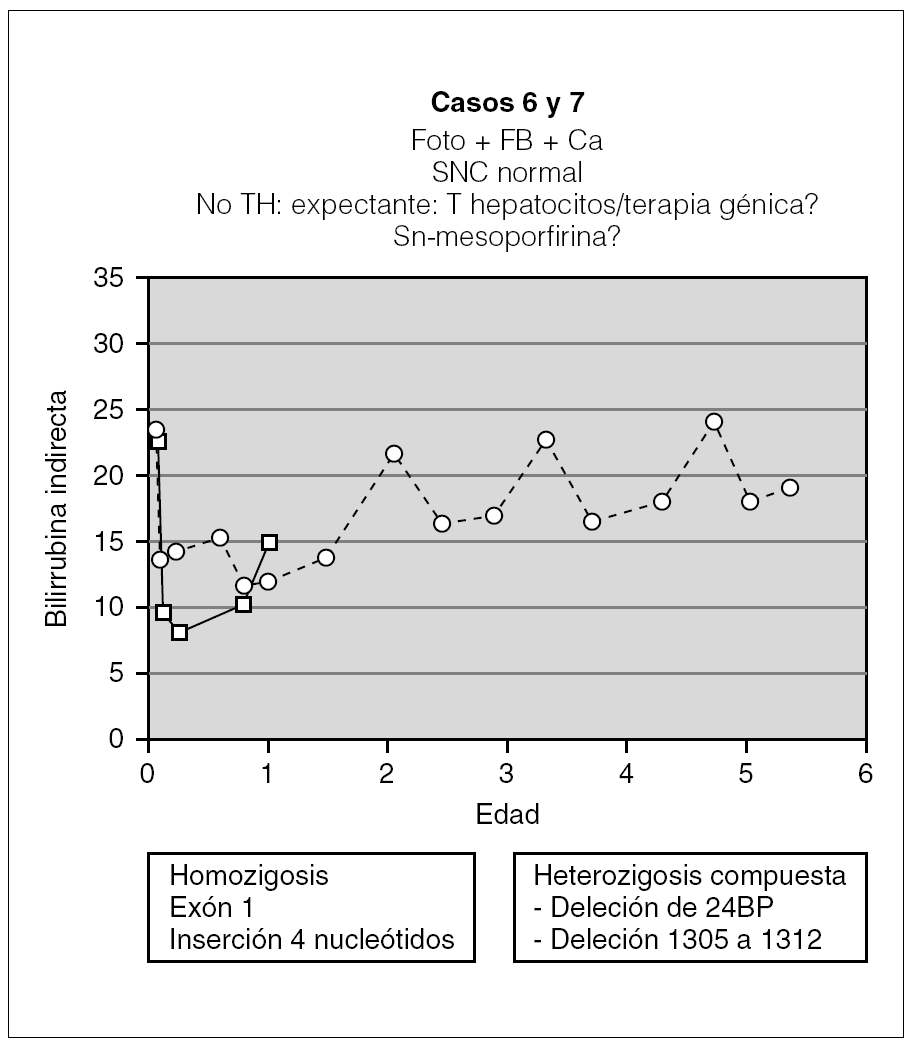
Los 2 pacientes restantes tienen ahora 2 y 6 años, y fueron diagnosticados a los 60 días y a la semana de vida, respectivamente. Ambos siguen tratamiento con fototerapia entre 9 y 16 h al día y con quelantes de sales biliares. Con ello mantienen la BI < 25 mg/dl (fig. 5) con desarrollo neurológico dentro de la normalidad. A pesar de la buena evolución en ambos casos, conviene mantener una actitud expectante en el futuro, ya que con la edad la fototerapia parece ser menos efectiva y estos pacientes son los más jóvenes de la serie.
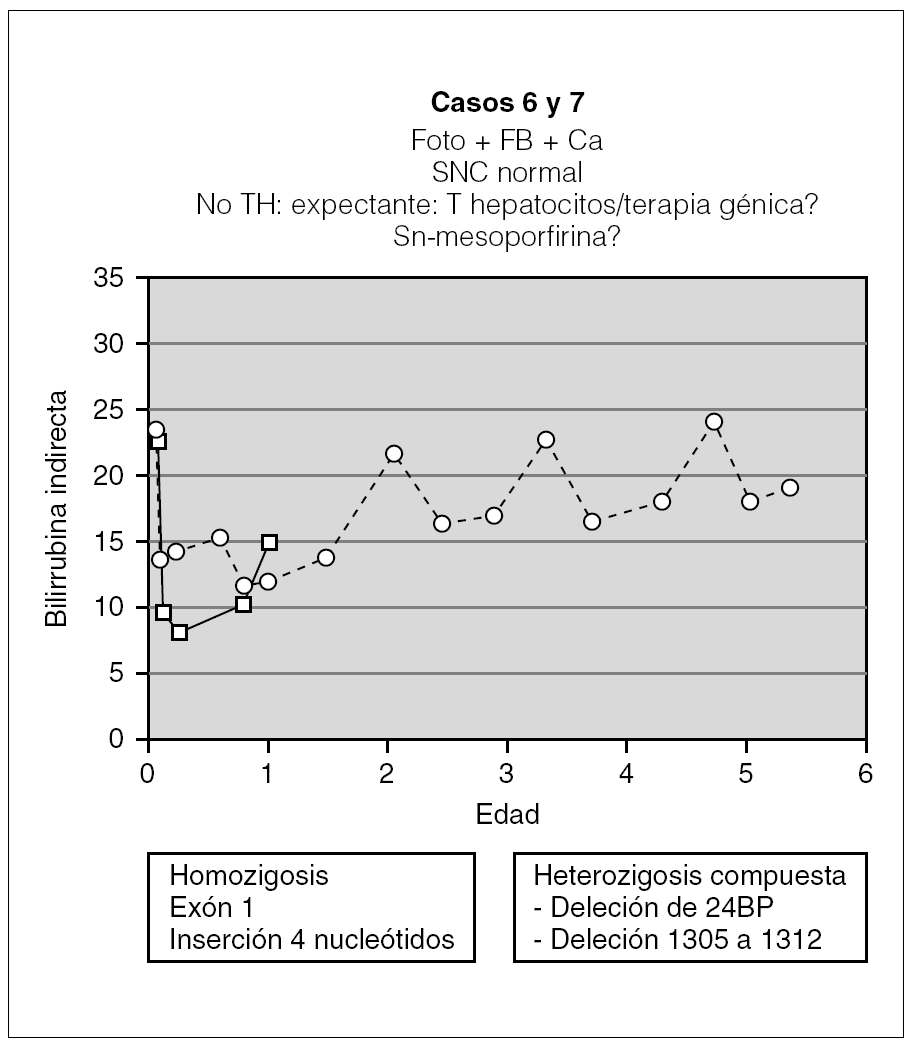
Figura 5. Evolución de los 2 pacientes con actitud expectante (foto: fototerapia).
Discusión
El hecho de que el SCN y el síndrome de Gilbert estén producidos por alteraciones del gen UGT1A1 explica la elevada frecuencia de antecedentes familiares con este síndrome entre los pacientes con SCN (3 de nuestros 7 niños). Por otra parte, el hallazgo entre los 7 casos de mutaciones no descritas hasta entonces puede estar justificado por los pocos pacientes diagnosticados hasta la actualidad, de modo que aún no se han encontrado la gran mayoría de las mutaciones posibles.
La sospecha diagnóstica de esta enfermedad debe hacerse lo más precozmente posible ante cualquier neonato con hiperbilirrubinemia indirecta sin signos de hemólisis y con función hepática normal. La confirmación se realizará posteriormente con el análisis genético 2,3,6, aunque también se puede demostrar la ausencia total de la enzima en tejido hepático o la inexistencia de bilirrubina conjugada en el aspirado duodenal 8,9.
El tratamiento inicial se fundamenta en la fototerapia 13, que se aplica durante un número de horas variable, que en los pacientes de nuestro estudio fue de un mínimo de 6 h diarias. Este tratamiento es menos efectivo a partir de la pubertad debido al aumento de grosor y de pigmentación de la piel y a la disminución de la superficie en relación con la masa corporal total. A la fototerapia se le pueden añadir otros tratamientos como el fenobarbital, que es un inductor enzimático que favorece la acción de la UGT (teóricamente sólo sería útil en el SCN tipo 2), así como quelantes de las sales biliares (colestiramina, sales de calcio) que inhiben la reabsorción de la BI en el intestino disminuyendo la circulación enterohepática de la misma. Para disminuir rápidamente las cifras de BI durante una crisis se puede utilizar la plasmaféresis. No está claro por encima de qué concentración de BI en sangre se produce el daño neurológico responsable del querníctero, aunque parece que el riesgo se incrementa ante cifras superiores a 25 mg/dl 7.
El trasplante hepático, ya sea ortotópico o auxiliar, es actualmente el único tratamiento curativo 14,15. Como este tratamiento no está exento de complicaciones y también puede limitar la calidad de vida, es importante evaluar en cada paciente la indicación del mismo frente al riesgo de padecer querníctero. En general, se considera razonable trasplantar después de los 3 años de vida, ya que entonces las complicaciones del trasplante se reducen considerablemente, pero antes de la adolescencia, momento en que la fototerapia pierde eficacia.
Entre otras alternativas terapéuticas se podría encontrar la utilización de Sn-mesoporfirina, que es un análogo estructural del grupo hemo capaz de bloquear el lugar de la hemooxigenasa en el que se inicia la conversión del hemo a bilirrubina 16. La porción del hemo cuya conversión a bilirrubina queda así interrumpida no se acumula en los tejidos, sino que se excreta intacta a través del sistema biliar. No se ha demostrado todavía la eficacia de este fármaco debido a que existe poca experiencia. El trasplante de hepatocitos 17-19 es una nueva opción de tratamiento que frente al trasplante hepático conlleva menos riesgos, ya que garantiza la función hepática en caso de rechazo de las células injertadas y evita el problema de la falta de donantes, pero, por el contrario, no elimina la necesidad de inmunosupresión y sólo supone una mejoría parcial de la enfermedad sin que existan evidencias claras sobre la funcionalidad de las células a largo plazo. Por último, otras técnicas de terapia génica, a pesar de ser esperanzadoras, sólo han sido llevadas a cabo de forma experimental en el tratamiento de diversas enfermedades genéticas.
En conclusión, se puede decir que esta rara enfermedad tiene un tratamiento difícil por cuanto todos entrañan riesgos importantes y disminuyen la calidad de vida de los pacientes. Es importante hacer un análisis individualizado encaminado a mantener cifras de BI no superiores a 25 mg/dl para reducir las probabilidades de querníctero.
Correspondencia: Dra. B. Lodoso Torrecilla.
Colombia, 25, 1.º A. 28016 Madrid. España.
Correo electrónico: blodoso@hotmail.com
Recibido en julio de 2005.
Aceptado para su publicación en febrero de 2006.
