




El síncope es un fenómeno común en la edad pediátrica, de origen mayoritariamente benigno1. A pesar de que la anamnesis y el examen objetivo a menudo permiten excluir situaciones de mayor gravedad, actualmente se recomienda la realización de un electrocardiograma (ECG) al constatar un primer episodio, esencial para la identificación de arritmias que, aunque raras, pueden ser potencialmente muy graves2. La prolongación del intervalo QT (QTc>044) es un ejemplo de arritmia que puede ocasionar síncope y muerte prematura. Puede ser congénita (síndrome de QT largo), resultante de alteraciones de los canales de sodio y de potasio de la membrana de las células miocárdicas, o adquirida, mediada por medicamentos o drogas, isquemia miocárdica, neuropatía autonómica, hipotiroidismo, accidentes cerebrovasculares y alteraciones electrolíticas, especialmente hipocaliemia, hipomagnesemia y, con menos frecuencia, hipocalcemia3.
Se presenta el caso de un adolescente de sexo masculino, de 15 años de edad, con múltiples ingresos por síncope. Los episodios de pérdida de consciencia eran descritos como breves, sin pródromo ni movimientos de los miembros o incontinencia, y no tenían relación aparente con situaciones de estrés o mayor actividad física. Se rechazaron antecedentes patológicos relevantes y el consumo de medicamentos o drogas. El examen objetivo era normal. Fue orientado a consulta de Cardiología, donde se diagnosticó prolongación del intervalo QTc en lo ECG (512ms), iniciándose tratamiento betabloqueador (atenolol, 100mg/día).
Cerca de 2 meses después, tenía un nuevo síncope y el ECG mostraba persistencia de la prolongación del intervalo QT (fig. 1). Se efectuó un estudio analítico que reveló hipocalcemia (calcio total 4,8mg/dl [N 8,4 a 10,2]; calcio ionizado 0,31mmol/l [N 1,2 a 1,38]); hiperfosfatemia (77,4mg/l [N 27 a 45]) e hipomagnesemia (1,42 mEq/l [N 1,55 a 2,05]), estando dentro de la normalidad los restantes parámetros analizados.
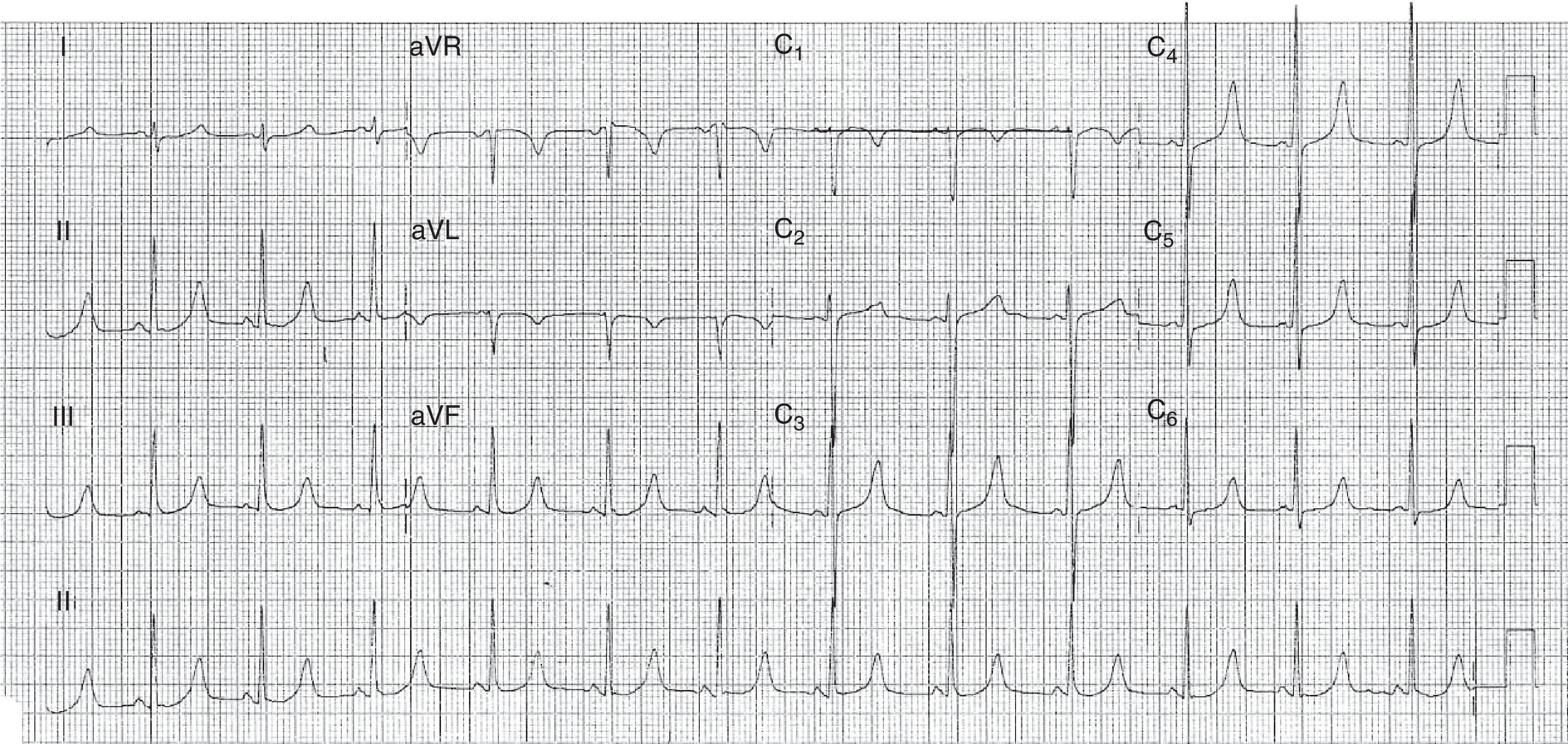
Con la administración por vía intravenosa de calcio y magnesio, y después de calcio, magnesio y calcitriol por vía oral, se experimentó una progresiva reducción del intervalo QT. El estudio complementario mostró un marcado aumento del valor de parathormona (PTH) (357pg/ml [N –10 a 65]), con función renal y dosificación de 1,25-dihidroxivitamina D normales. La conjugación de hipocalcemia, hiperfosfatemia y PTH elevadas sugería seudohipoparatiroidismo (PHP).
La PTH es responsable del mantenimiento del equilibrio fosfocálcico y cuando esta hormona se produce de forma insuficiente (hipoparatiroidismo) o hay resistencia a su acción en los órganos objetivo (PHP), existe una reducción significativa de los niveles plasmáticos de calcio y un aumento de los niveles de fósforo4,5. En el PHP, los valores de PTH están elevados por hiperproducción refleja6.
El PHP es una enfermedad rara y se deriva de alteraciones de la transducción de la señal tras la conexión de la PTH a sus receptores4. Dependiendo de las mutaciones o las modificaciones del imprinting que afectan a las vías de transducción de señal, pueden surgir diferentes subtipos de PHP: Ia, Ib, Ic y II5,7 (tabla 1).
Diagnóstico diferencial de los diferentes tipos de seudohipoparatiroidismo
| Calcio | Fósforo | PTH | OHA | Resistencia plurihormonal | ↑ AMPc urinario tras PTH | Actividad Gsα | |
| PHP Ia | ↓ | ↑ | ↑ | Sí | Sí | No | Alterada-mutación GNAS |
| PHP Ib | ↓ | ↑ | ↑ | No | No | No | Alterada-alteración de la metilación GNAS |
| PHP Ic | ↓ | ↑ | ↑ | Sí | Sí | No | N |
| PHP II | ↓ | ↑ | ↑ | No | No | Sí | N |
| PPHP | N | N | N | Sí | No | Sí | Alterada-mutación GNAS |
AMPc: monofosfato cíclico de adenosina; Gsα: subunidad α de la proteína G estimuladora; N: normal; OHA: osteodistrofia hereditaria de Albright; PHP: seudohipoparatiroidismo; PPHP: seudoseudohipoparatiroidismo; PTH: parathormona.
Los pacientes con PHP Ia suelen presentar una constelación de características fenotípicas (baja estatura, cara redonda, braquidactilia, obesidad, osificaciones ectópicas y, en ocasiones, retraso mental), que conjuntamente se denominan osteodistrofia hereditaria de Albright (OHA)7. En el PHP Ib la resistencia hormonal parece circunscribirse a la acción de la PTH en los túbulos proximales renales y, por eso, los pacientes no presentan las alteraciones fenotípicas de OHA8-10. El diagnóstico del PHP tipo Ib suele ser tardío, ya que a menudo las manifestaciones surgen únicamente durante la pubertad, estadio en que aumentan las necesidades de calcio. El PHP Ic es una forma más rara, caracterizada por la presencia de OHA y resistencia hormonal múltiple4,7. En el PHP II, como las alteraciones de transducción de la señal son distales a la producción del AMPc, se produce un aumento del AMPc tras la administración exógena de PTH7.
En el paciente presentado no eran evidentes alteraciones que sugirieran OHA, la dosificación de las restantes hormonas era normal y no se observó elevación del AMPc urinario. De este modo, se planteó la sospecha de que podía tratarse del subtipo Ib, solicitándose la evaluación del patrón de metilación del gen GNAS, que mostró la ausencia de metilación del exón A/B del alelo materno, confirmando el diagnóstico de PHP tipo Ib.
El tratamiento de la hipocalcemia grave debe efectuarse con administración por vía intravenosa de calcio y, si fuera necesario, también deben ser corregidas la hipomagnesemia, la hiperfosfatemia y la alcalosis4. A largo plazo, se administra calcio por vía oral y vitamina D activa y se recomienda la vigilancia bioquímica (calcio, fósforo, PTH, creatinina) y urinaria (calcio, creatinina) a cada 3 meses y el estudio radiológico regular para exclusión de nefrocalcinosis4,5,9.
