

Introducción
El diagnóstico de un recién nacido (RN) con genitales ambiguos es una urgencia de difícil manejo. A la incertidumbre y ansiedad familiar originada al plantear dudas sobre el sexo del RN, se suma que el diagnóstico médico es en ocasiones complicado y requiere una actuación multidisciplinaria: pediatra, genetista, analista, radiólogo y cirujano infantil. Dentro de las heterogéneas causas de ambigüedad de los genitales externos, el seudohermafroditismo masculino (SHM) conlleva una mayor complejidad ya que, a diferencia del femenino, cuya causa más frecuente es la hiperplasia suprarrenal congénita (HSC) de fácil diagnóstico en la mayoría de las ocasiones, el masculino requiere agotar gran parte de los recursos disponibles (determinaciones hormonales, estudios genéticos, de imagen, moleculares, etc.), al no aportar la clínica una orientación diagnóstica clara. Presentamos 2 casos de SHM, repasando las posibles causas que lo producen así como las pruebas complementarias necesarias para su diagnóstico.
Observaciones clínicas
Caso 1
Recién nacido a término de 2.825 g que ingresó para estudio de genitales ambiguos ya observados en ecografías prenatales (hipertrofia de clítoris). Antecedentes perinatales sin interés. Exploración con genitales ambiguos en estadio 3 de Prader (clítoris hipertrófico, labios mayores normales no fusionados, uretra y vagina terminando en un conducto común). Se realizaron: cariotipo 46XY, testosterona 1,33 ng/ml (0,2-1,7), hormona foliculoestimulante (FSH) 10,5 mUI/ml (0,5-4,5), hormona luteinizante (LH) 18 mUI/ml (0,5-2,5). 17-OH progesterona: normal. Test de estímulo con gonadotropina coriónica humana (HCG) (500 U/48 h, 7 dosis): determinaciones basales: testosterona (TST), 1,85 ng/ml (0,2-1,7); delta-4 androstendiona, 1,6 ng/ml (0,5-2,5); dihidrotestosterona (DHT), 0,56 ng/ml (0,09-0,13); cociente TST/DHT: 3,3 (12,3-15,5). Determinaciones tras estímulo: TST: 1,92 ng/ml (1-6); delta-4 androstendiona: 1,3 ng/ml; DHT: 0,43 ng/ml; cociente TST/DHT: 4,4 (20-250); 17-OH progesterona: 1,45 ng/ml (< 20). Ecografía abdominopélvica: se visualizó una masa de aspecto uterino. Cistografía/genitografía: vejiga normal sin reflujo vesicoureteral. Imagen triangular posterior a la vejiga sugestiva de vagina. Estudio del gen del receptor de andrógenos normal. La resonancia mostró un útero sin visualización de gónadas. En la laparoscopia exploradora se visualizaron útero y trompas normales, con gónadas de aspecto macroscópico normal. No se visualizaron conductos deferentes. Se realizó sinuscopia observándose vagina y cuello de útero normales. El estudio anatomopatológico mostró en gónada derecha un ovoteste con trompa hipoplásica y epidídimo y en gónada izquierda una cintilla fibrosa lo que confirmó el diagnóstico de hermafroditismo verdadero (HV). Se asignó sexo femenino, se procedió a gonadectomía y se decidió cirugía correctora al año de vida.
Caso 2
Recién nacido a término de 2.650 g sin antecedentes perinatales y familiares destacables, con padres jóvenes no consanguíneos. Exploración con una arteria umbilical única, y la existencia de un tubérculo genital de 1,5 cm con una hendidura genital de 1 a 1,5 cm de longitud en su base, pliegues labioescrotales lisos en cuyo interior se palpaban 2 bultomas de consistencia elástica de 1 x 1 cm. No se apreció orificio vaginal (estadio 3-4 de Prader). Se realizaron: cariotipo 46XY; 17-OH progesterona: 17,8 ng/ml (< 25); androstendiona: 1,7 ng/ml (0,5-2,5); FSH: 0,98 mUI/ml (0,5-4,5); LH: 7,09 mUI/ml (0,5-2,5); cortisol: 49,06 ng/ml (50-200). Test de estímulo con HCG a los 20 días de vida (determinaciones basales: TST: 2,57 [0,2-1,7] ng/ml; DHT: no realizada). Determinaciones tras estímulo: TST, 6,15 ng/ml; DHT: 0,4 ng/ml; cociente TST/DHT: 15,3 (20-250). Ecografía abdominopélvica: testes y epidídimo por debajo del canal inguinal, con tamaño y ecoestructura dentro de la normalidad. CUMS: uretra de morfología femenina con meato en base de pene, sin impronta del verum montarum, ni utrículo prostático. Estudio molecular para el gen del receptor de andrógenos normal. Estudio para el gen de la 5a -reductasa tipo 2 (SRD5A2), con presencia de 2 mutaciones (Q126R en el exón 2 y A207D en el exón 4), diagnosticándose de SHM por déficit de la enzima 5a -reductasa. Cada padre era portador de una de las mutaciones. Se le asignó sexo masculino decidiéndose cirugía correctora en dos tiempos al año de vida. Recibió tratamiento con testosterona intramuscular a dosis de 50 mg/mes durante 4 meses observándose un incremento del tamaño del pene de 1,5 a 2,2 cm, continuándose posteriormente con dihidrotestosterona tópica al 2 % durante un mes.
Discusión
El dimorfismo sexual es el resultado de complejos mecanismos que definen la diferenciación sexual, incluyendo la determinación del sexo genético, la diferenciación gonadal y genital, así como el desarrollo psicosexual1. En la ambigüedad genital, el aspecto de los genitales externos no permite una asignación inequívoca del RN a un sexo u otro. Es una urgencia neonatal, ya que pueden incluirse entidades con síndrome pierde sal como la HSC. Debe asignarse el sexo al RN antes de que fije su sexo psicológico, con las implicaciones sociales y legales derivadas2,3. El SHM es la situación en la que un individuo 46XY con tejido testicular en sus gónadas, presenta una virilización incompleta de sus genitales internos y/o externos, con aparición de unos genitales externos ambiguos o claramente femeninos. Clásicamente se han distinguido tres niveles de diferenciación sexual: el sexo genético, el gonadal y el genital, existiendo en el caso del SHM una ausencia de correspondencia entre ellos4. Todos quedan determinados durante el período fetal. Para la diferenciación de la gónada primitiva hacia testículo es imprescindible la expresión de una cascada de genes, uno de los cuales está localizado en el cromosoma Y (fig. 1). Una vez diferenciada hacia testículo, el proceso de diferenciación genital masculino requiere la expresión de una serie de genes, cuyo fallo provoca diferentes tipos de SHM5.
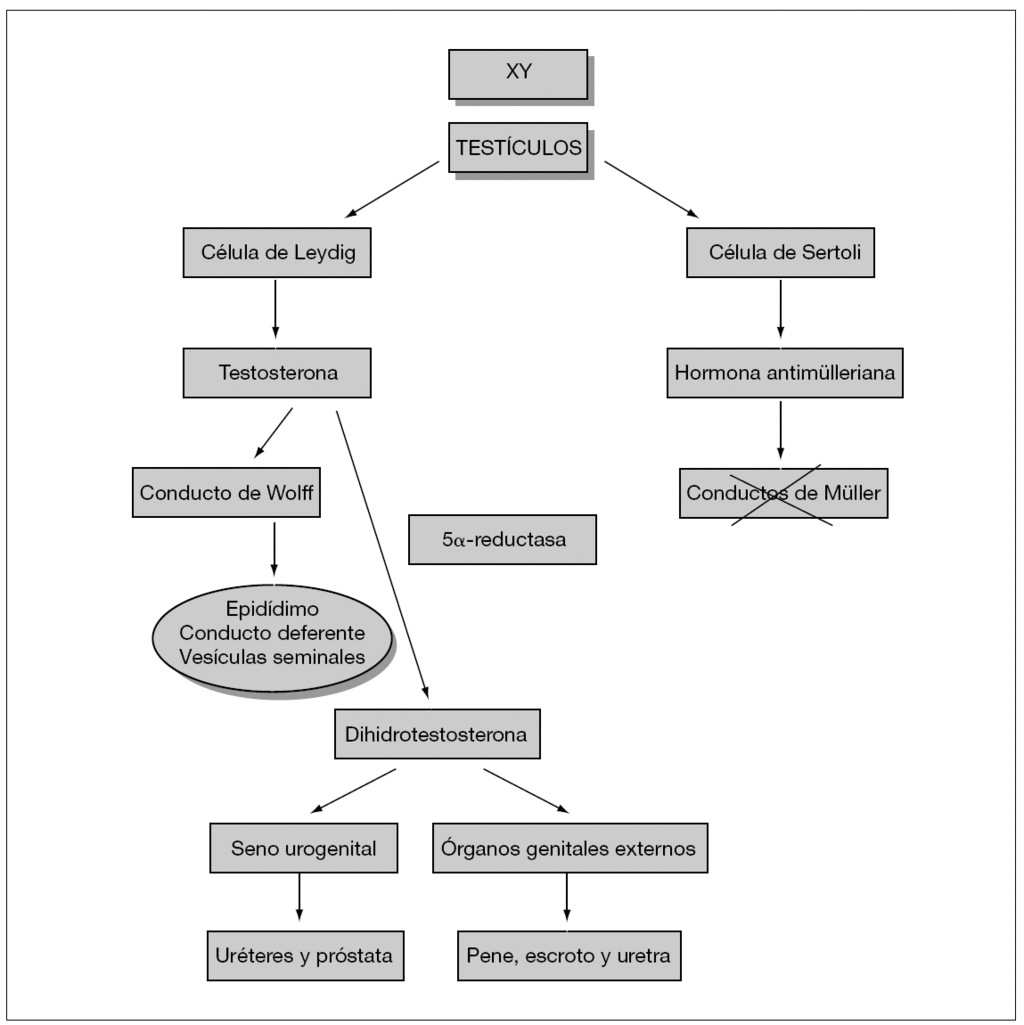
Figura 1. Diferenciación sexual.
El fallo androgénico con hormona antimülleriana normal es la situación más frecuente incluyendo varios procesos. La ausencia de respuesta testicular a la LH puede deberse bien a alteraciones moleculares de la misma o a anomalías de su receptor. En ambos casos se produciría una aplasia/hipoplasia de las células de Leydig. Los valores plasmáticos de testosterona se encontrarían disminuidos, con niveles elevados de LH. La respuesta de la TST al estímulo con HCG nos permite diferenciar ambas entidades. No habría respuesta si la anomalía estuviera en el receptor de la LH.
El déficit en la síntesis de TST se debe a errores congénitos en su producción, secundarios a defectos enzimáticos (fig. 2). Son raros, de herencia autosómica recesiva y suponen el 10 % de los casos de SHM6. De las cuatro enzimas que intervienen en la síntesis de testosterona, sólo la 17-ceto-reductasa es exclusivamente gonadal. El déficit de las tres primeras produciría cuadros de HSC cuyo diagnóstico se basa en determinaciones basales y tras estímulo con ACTH. En el déficit de 17-ceto-reductasa se observa un aumento de androstendiona tras estímulo con HCG, con niveles de TST bajos y sin respuesta tras el mismo.
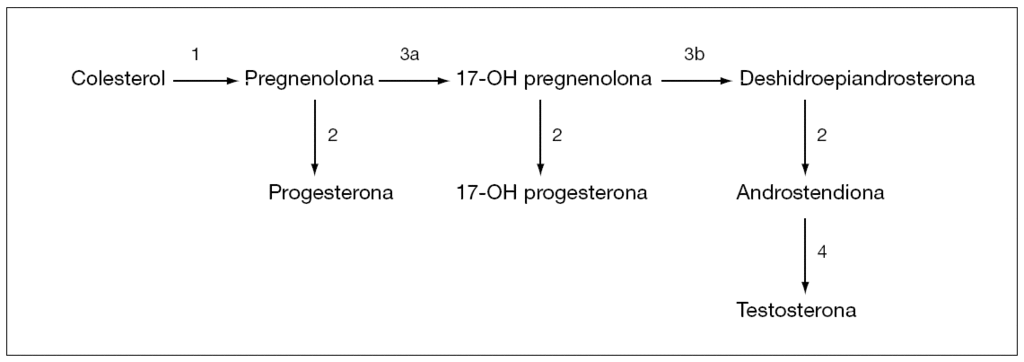
Figura 2. Síntesis de testosterona. 1: P450scc; 2: 3bhidroxiesteroide deshidrogenasa; 3: P450c17 con actividad hidroxilasa (3a) y con actividad desmolasa (3b); 4: 17b hidroxiesteroide deshidrogenasa.
El déficit de receptores de andrógenos, anteriormente conocido como síndrome de Morris o de feminización testicular, es la causa más frecuente de SHM (60-70 % de los casos). Su denominación actual es síndrome de insensibilidad a los andrógenos (AIS) en su forma completa (CAIS) o parcial (PAIS). Los niveles basales de testosterona y dihidrotestosterona se encuentran en el límite alto de la normalidad o ligeramente elevados. El diagnóstico se basa en el estudio del gen que codifica dicho receptor, situado en el brazo largo del cromosoma X. Hay descritas gran número de mutaciones y anomalías moleculares.
El déficit de conversión de TST a DHT se debe a un déficit de la enzima 5α -reductasa tipo 2. Los niveles basales de TST serían normales o altos, mientras que los de DHT se encontrarían bajos, elevándose el cociente TST/DHT. Tras estímulo con HCG, este cociente se multiplicaría por 3 o 4. Se puede estudiar el gen que codifica dicha enzima (SRD5A2), situado en el cromosoma 2, habiéndose descrito distintas mutaciones. De herencia autosómica recesiva afecta exclusivamente a varones. Existen dos isoformas distintas de la 5α -reductasa: la 1, codificada por un gen en el cromosoma 5 y que se expresa en el hígado y folículo pilosebáceo y la 2, codificada por un gen en el cromosoma 2 y que se expresa en la próstata y genitales externos. En la mayoría de los casos, la sintomatología se debe a mutaciones en la isoenzima 27. La asignación de sexo masculino es importante, ya que en la pubertad va a producirse una importante virilización del paciente, debido a la falta de retroalimentación negativa de la DHT sobre la hipófisis, provocando un aumento de LH, del cociente TST/DHT y, merced a la acción de la isoenzima 1, de la DHT8,9.
En el fallo de la hormona antimülleriana, por alteración en su producción, liberación o acción, con producción androgénica normal, los pacientes presentan genitales externos no ambiguos, totalmente virilizados pero con restos de conductos de Müller (suelen diagnosticarse al operar una hernia inguinal en la que se encuentra el útero, trompas y parte superior de vagina).
Fallan ambas hormonas, antimülleriana y andrógenos, en los casos de disgenesia gonadal con un desarrollo anormal de la gónada cuyo tejido es sustituido por tejido fibroso no funcionante. Los genitales internos pueden variar desde totalmente femeninos (disgenesia gonadal pura XY), a ser una combinación de derivados müllerianos y wolfianos (disgenesia gonadal parcial XY, SHM disgenético). Los genitales externos presentan también diversos grados de ambigüedad. Hay que hacer el diagnóstico diferencial con el hermafroditismo verdadero (coexisten parénquima testicular y ovárico), requiriéndose el estudio anatomopatológico de la gónada.
En el caso 1, la ausencia de bultomas nos indujo a pensar en un seudohermafroditismo femenino cuya causa más frecuente es la HSC. La ausencia de hiperpigmentación genital y la normalidad de la 17-OH progesterona la descartaron. El cariotipo delimitó las posibilidades hacia un SHM. Los niveles algo elevados de gonadotropinas y testosterona, sugirieron la posibilidad de una insensibilidad a los andrógenos no confirmada posteriormente por estudios moleculares. El test de estímulo con HCG (escasa elevación de TST y DHT, permaneciendo el cociente TST/DHT en límites normales) descartó el déficit de 5α -reductasa. Los niveles normales de androstendiona tras HCG también descartaron el déficit de 17-ceto-reductasa. La presencia de derivados müllerianos, orientó hacia una disgenesia gonadal o hacia un HV, siendo este último el diagnóstico definitivo. Menos de un 10 % de estos últimos presentan un cariotipo 46XY, siendo lo más habitual el cariotipo 46XX (70-80 %) o un mosaicismo (10 %)10.
En el caso 2 la presencia de bultomas nos indujo a pensar en un SHM. Tras el test de estímulo con HCG, el cociente TST/DHT elevado sugirió un déficit de 5α -reductasa confirmado con el estudio genético.
El diagnóstico de una ambigüedad sexual puede ser fácil conociendo la fisiopatología de la diferenciación gonadal y genital. Una meticulosa exploración física es fundamental, siendo de gran valor la presencia o no de bultomas en región inguinal o formaciones labioescrotales. Su presencia casi siempre sugiere la existencia de testículos o más raramente de ovotestes lo que supondría un SHM o un HV. Con un cariotipo, determinaciones hormonales basales y tras estímulo, factor antimülleriano (que significa presencia de tejido testicular) y una ecografía puede conseguirse una orientación diagnóstica bastante aproximada, completándose con estudios moleculares o de imagen más complejos11,12. Recientemente se ha propuesto una nueva nomenclatura en el campo de los trastornos de la diferenciación sexual, en la cual los SHM pasarían a ser denominados "Intersexo 46XY poco virilizado"13.
Correspondencia: Dr. L. Tapia Ceballos.
Departamento de Pediatría. Hospital Costa del Sol.
Ctra. Nacional 340, km 187. 29600 Marbella. Málaga. España.
Correo electrónico: leotapiaceb@hotmail.com
Recibido en agosto de 2006.
Aceptado para su publicación en marzo de 2007.
