
Con las terapias actuales, la supervivencia de los tumores del sistema nervioso central (TSNC) es cada vez mayor y, con ello, las complicaciones a largo plazo.
ObjetivoEvaluar las secuelas endocrinológicas en niños con TSNC en relación con el tipo de neoplasia y el tratamiento recibido.
Sujetos y métodosSe revisaron retrospectivamente los datos clínicos, auxológicos, analíticos y radiológicos de 38 pacientes (36,8% mujeres y 63,2% varones) con antecedente de TSNC y seguimiento mínimo de 5 años.
ResultadosLa media±desviación estándar de edad al diagnóstico fue de 5,34±3,07 años. El 76,3% de los casos presentó al menos un déficit hormonal, siendo el más prevalente el de hormona de crecimiento (GH) (73,7%), seguido de los déficits de tirotropina (TSH) (68,4%), corticotropina (31,6%), hormona antidiurética (28,9%) y gonadotropinas (LH/FSH) (21,1%). El 21,1% de los pacientes presentaron pubertad precoz. A los 5 años de seguimiento, el 28,9% presentaba obesidad. El craneofaringioma fue el tipo tumoral que registró mayor número de casos con deficiencias hormonales, obesidad y tasa de recurrencia. El tratamiento más frecuentemente administrado fue la combinación de cirugía+quimioterapia+radioterapia, empleado en el 47,4% de los pacientes. La talla final media ± desviación estándar (20 pacientes) fue −1,2±1,6; con una disminución media de −0,53 DE respecto de su talla diana.
Conclusiones1) El tipo tumoral y el tratamiento recibido influyen sobre las secuelas endocrinológicas; 2) las deficiencias hormonales más frecuentes de todos los tipos de TSNC, independientemente del tratamiento recibido, fueron GH y TSH; 3) el diagnóstico precoz y la intervención temprana sobre la disfunción endocrina, reducen la morbilidad y mejoran la calidad de vida a largo plazo.
Given the successful increase in survival rates with the current treatments for central nervous system tumours (CNST), survivors are at high risk for late adverse effects.
PurposeTo evaluate the endocrine sequelae in children with CNST according to the type of tumour and treatment received.
Patients and methodsA retrospective review of the clinical features, auxology, hormone determinations and imaging findings of 38 patients (36.8% females, 63.2% males) with CNST, with a minimum of 5 years follow-up, was performed.
ResultsThe mean age at diagnosis was 5.34±3.07 years, with 76.3% of the patients having at least one hormone deficiency, of which growth hormone (GH) (73.7% of all patients) was the most prevalent, followed by thyrotropin (TSH) (68.4%), corticotropin (31.6%), antidiuretic hormone (28.9%), and gonadotropin (LH/FSH) (21.1%) deficiency. Precocious puberty was found in 21.1% of patients. After 5 years of follow-up, 28.9% were obese. Craniopharyngioma had more hormone deficiencies, obesity and recurrence rates. The most frequently administered treatment was surgery + chemotherapy + radiotherapy, in 47.4% of the patients. Mean final height (20 patients) was −1.2 1.6 SDS, with a mean difference of −0.53 SDS regarding their target height.
Conclusions1) The type of tumour and treatment received influence the endocrinological sequelae. 2) The most frequent hormone deficiencies in all types of CNST, regardless of the treatment received, were GH and TSH. 3) Early diagnosis and prompt intervention of endocrine dysfunction can reduce the morbidity and improve quality of life over the long term.
En la infancia, los tumores del sistema nervioso central (TSNC) constituyen el tipo de neoplasia sólida más frecuente y la segunda causa de neoplasia maligna (20-25% del total1) tras las hematológicas2. La incidencia de los TSNC en nuestro medio es 3/100.0001 en menores de 15 años, con una relación varón/mujer de 1,22.
Su localización predominante en los 2 primeros años de vida es supratentorial; en el resto de la primera década, infratentorial, para volver a predominar la supratentorial en la adolescencia y la edad adulta2. Los tipos histológicos más frecuentes son: astrocitoma (24%), glioma (22%), meduloblastoma/tumores neuroectodérmicos primitivos (10%), tumores hipofisarios y craneofaringioma (10%), ependimoma (6%) y tumores germinales (4%)1.
Para la mayoría de los TSNC, el tratamiento incluye la cirugía, salvo para aquellos mal circunscritos o sin posibilidad de resección. La mortalidad quirúrgica en manos expertas es del 1%3 y su morbilidad varía según la localización tumoral y la terapia adyuvante3. La indicación de radioterapia depende de la histología tumoral2 y puede administrarse focalmente (sobre la lesión) u holocraneal+espinal+refuerzo sobre la lesión3. Los TSNC pediátricos precisan dosis altas de radioterapia, típicamente>30Gy (frecuentemente 50-60Gy)3. La edad es un factor de riesgo para desarrollar complicaciones a largo plazo por radioterapia; apareciendo alteración intelectual profunda en aquellos que reciben radiación antes de los 3 años de edad4. El empleo de quimioterapia en el tratamiento de los TSNC ha aumentado en los últimos años, convirtiéndose en estándar para evitar la radioterapia en menores de 3 años5. Los agentes empleados dependen de la sensibilidad de cada tipo tumoral.
Con los protocolos terapéuticos actuales, la supervivencia de los TSNC es cada vez mayor (73,3% a 5 años6,7) y con ello las secuelas endocrinas asociadas a largo plazo (43% de ellas)8. Estos efectos se relacionan con el efecto directo del tumor, su localización, extensión-infiltración de otras estructuras, tratamientos recibidos, edad al diagnóstico, sexo y tiempo transcurrido desde la finalización del tratamiento6. Debido a la vulnerabilidad y las limitadas propiedades reparativas del tejido cerebral, los supervivientes de TSNC tienen un riesgo elevado de efectos adversos9,3. Se define efecto a largo plazo como cualquier consecuencia física, médica, cognitiva o psicosocial crónica que acontece pasados 5 años tras el diagnóstico de un tumor. El establecimiento de la cifra en 5 años10 obedece a que este suele ser el tiempo considerado para definir supervivencia al cáncer11.
ObjetivoEvaluar las tasas de secuelas endocrinológicas en niños con TSNC seguidos en el Servicio de Endocrinología del Hospital Infantil Universitario Niño Jesús de Madrid, según el tipo de neoplasia y el tratamiento recibido.
Sujetos y métodosSe revisaron de forma retrospectiva, entre los meses de diciembre del 2011 y febrero del 2012, las historias clínicas de los pacientes con TSNC, con un seguimiento mínimo de 5 años.
Las alteraciones endocrinas fueron determinadas mediante las siguientes evaluaciones:
Clínica y auxológica- –
Monitorización del crecimiento: peso, talla e índice de masa corporal (IMC) en el momento del diagnóstico y semestralmente hasta alcanzar la talla adulta.
- –
Exploración física: general, puberal y tiroidea al diagnóstico y en cada consulta hasta completar su desarrollo puberal.
- –
Determinación anual de insulin-like growth factor-1 (IGF-1), insulin-like growth factor binding protein-3 (IGFBP-3), perfil tiroideo (TSH y T4 libre), prolactina, cortisol matinal y testosterona/estradiol (este último según estadio puberal). Asimismo, se realizaron pruebas de estimulación de hormona de crecimiento (GH) (clonidina e hipoglucemia insulínica), de gonadotropinas (FSH, LH) (estimulación con hormona liberadora de gonadotrofinas) y corticotropina (test de ACTH) ante sospecha de deficiencia de dichas hormonas.
- –
Pruebas de imagen: edad ósea anual. Ecografía tiroidea anual en caso de radiación craneal/espinal. Resonancia magnética craneal de acuerdo con el equipo oncológico. Ecografía pélvica para valorar inicio/progresión puberal o en pacientes que recibieron radioterapia espinal o quimioterapia gonadotóxica.
El estudio incluye a 38 pacientes con una media ± desviación estándar (DE) de edad al diagnóstico de 5,34±3,07 años. La distribución por sexo fue: 36,8% mujeres y 63,2% varones.
Los tipos tumorales analizados y su frecuencia fueron: 26,3% (n=10) meduloblastomas, 21,1% (n=8) craneofaringiomas, 18,4% (n=7) astrocitomas, 10,5% (n=4) germinomas, 10,5% (n=4) ependimomas, 2,6% (n=1) gliomas y 10,5% (n=4) otros tipos. Los 4 casos de «otros tipos tumorales» corresponden a 2 hamartomas y 2 rabdomiosarcomas.
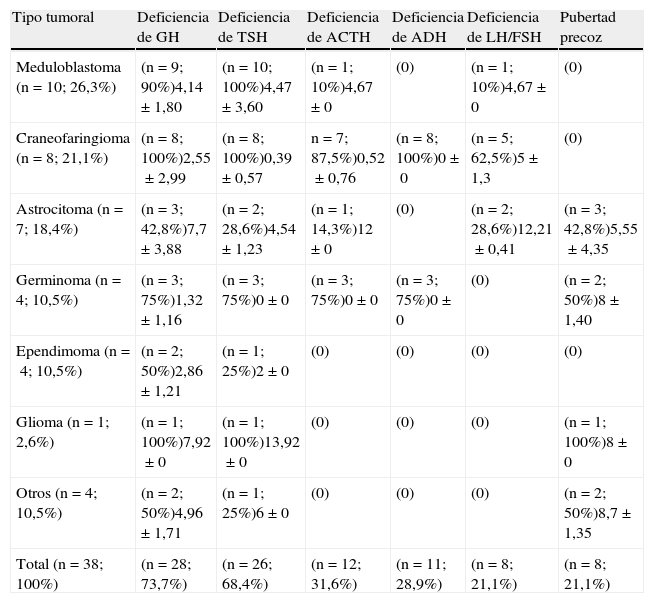
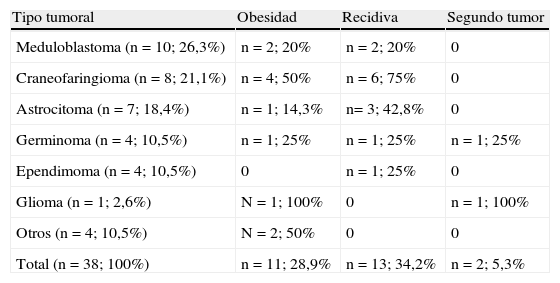
Entre los pacientes analizados, el 76,3% presentó al menos un déficit hormonal. El déficit más prevalente fue el de GH con el 73,7% de los casos, seguido de los déficits de TSH (68,4%), ACTH (31,6%) y LH/FSH (21,1%). El 21,1% (n=8) de estos pacientes presentó pubertad precoz. A los 5 años de seguimiento, el 28,9% presentó obesidad (definida como IMC>2 DE de acuerdo con las referencias de Hernández et al., 1988). Se registró recidiva tumoral en el 34,2% (n=13) de los casos y segunda neoplasia en el 5,3% (n=2), sin producirse ningún fallecimiento durante el seguimiento. Las segundas neoplasias corresponden en el primer caso a un varón diagnosticado de glioma, que recibió tratamiento con cirugía+radioterapia craneal+quimioterapia y años después fue diagnosticado de carcinoma basocelular nodular en la región fronto-parietal. El otro caso corresponde a un varón diagnosticado de germinoma que recibió radioterapia craneal+quimioterapia, siendo años después diagnosticado de carcinoma papilar tiroideo. Las tablas 1 y 2 recogen los diferentes tipos tumorales, consecuencias endocrinas, recidiva y segundos tumores; así como el intervalo entre la administración del tratamiento y la alteración hormonal.
Tipos de tumores y secuelas endocrinológicas registradas. Se expresan el número y el porcentaje de casos en paréntesis, y la media de tiempo ± DE, expresado en años, entre el inicio del tratamiento y la alteración hormonal
| Tipo tumoral | Deficiencia de GH | Deficiencia de TSH | Deficiencia de ACTH | Deficiencia de ADH | Deficiencia de LH/FSH | Pubertad precoz |
| Meduloblastoma (n=10; 26,3%) | (n=9; 90%)4,14±1,80 | (n=10; 100%)4,47±3,60 | (n=1; 10%)4,67±0 | (0) | (n=1; 10%)4,67±0 | (0) |
| Craneofaringioma (n=8; 21,1%) | (n=8; 100%)2,55±2,99 | (n=8; 100%)0,39±0,57 | n=7; 87,5%)0,52±0,76 | (n=8; 100%)0±0 | (n=5; 62,5%)5±1,3 | (0) |
| Astrocitoma (n=7; 18,4%) | (n=3; 42,8%)7,7±3,88 | (n=2; 28,6%)4,54±1,23 | (n=1; 14,3%)12±0 | (0) | (n=2; 28,6%)12,21±0,41 | (n=3; 42,8%)5,55±4,35 |
| Germinoma (n=4; 10,5%) | (n=3; 75%)1,32±1,16 | (n=3; 75%)0±0 | (n=3; 75%)0±0 | (n=3; 75%)0±0 | (0) | (n=2; 50%)8±1,40 |
| Ependimoma (n=4; 10,5%) | (n=2; 50%)2,86±1,21 | (n=1; 25%)2±0 | (0) | (0) | (0) | (0) |
| Glioma (n=1; 2,6%) | (n=1; 100%)7,92±0 | (n=1; 100%)13,92±0 | (0) | (0) | (0) | (n=1; 100%)8±0 |
| Otros (n=4; 10,5%) | (n=2; 50%)4,96±1,71 | (n=1; 25%)6±0 | (0) | (0) | (0) | (n=2; 50%)8,7±1,35 |
| Total (n=38; 100%) | (n=28; 73,7%) | (n=26; 68,4%) | (n=12; 31,6%) | (n=11; 28,9%) | (n=8; 21,1%) | (n=8; 21,1%) |
Tipos de tumores y casos con obesidad (IMC>2 DE), recidiva y segundos tumores. Se expresa el número y porcentaje de casos registrados
| Tipo tumoral | Obesidad | Recidiva | Segundo tumor |
| Meduloblastoma (n=10; 26,3%) | n=2; 20% | n=2; 20% | 0 |
| Craneofaringioma (n=8; 21,1%) | n=4; 50% | n=6; 75% | 0 |
| Astrocitoma (n=7; 18,4%) | n=1; 14,3% | n= 3; 42,8% | 0 |
| Germinoma (n=4; 10,5%) | n=1; 25% | n=1; 25% | n=1; 25% |
| Ependimoma (n=4; 10,5%) | 0 | n=1; 25% | 0 |
| Glioma (n=1; 2,6%) | N=1; 100% | 0 | n=1; 100% |
| Otros (n=4; 10,5%) | N=2; 50% | 0 | 0 |
| Total (n=38; 100%) | n=11; 28,9% | n=13; 34,2% | n=2; 5,3% |
DE: desviación estándar; IMC: índice de masa corporal.
El craneofaringioma, dada su localización, fue el tipo tumoral que registró mayor número de casos con deficiencias hormonales, obesidad y tasa de recurrencia. Todos los craneofaringiomas (n=8) presentaron déficit de GH, TSH, ADH, y 7 de ellos además, déficit de ACTH y 5 casos hipogonadismo hipogonadotropo.
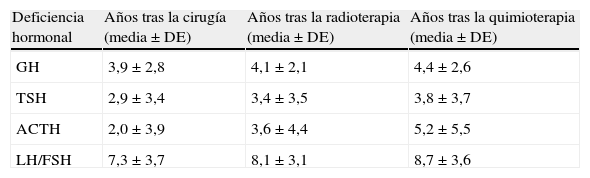
La tabla 3 muestra el tiempo medio entre la administración del tratamiento y la presentación de cada deficiencia hormonal. La administración de cirugía se sigue de la aparición de deficiencias hormonales hipofisarias (todos los tipos) más precoces en el tiempo que tras administrar radioterapia o quimioterapia. La deficiencia de ADH estaba presente en todos los pacientes (11 casos del total: 8 craneofaringiomas y 3 geminomas) desde su presentación clínica inicial.
Intervalo medio, expresado en años, entre el tratamiento y la presentación de deficiencia hormonal
| Deficiencia hormonal | Años tras la cirugía (media±DE) | Años tras la radioterapia (media±DE) | Años tras la quimioterapia (media±DE) |
| GH | 3,9±2,8 | 4,1±2,1 | 4,4±2,6 |
| TSH | 2,9±3,4 | 3,4±3,5 | 3,8±3,7 |
| ACTH | 2,0±3,9 | 3,6±4,4 | 5,2±5,5 |
| LH/FSH | 7,3±3,7 | 8,1±3,1 | 8,7±3,6 |
ACTH: hormona adrenocorticotropa o corticotropina; DE: desviación estándar; FSH: hormona foliculoestimulante; GH: hormona de crecimiento; LH: hormona luteoestimulante; TSH: hormona estimulante del tiroides o tirotropina.
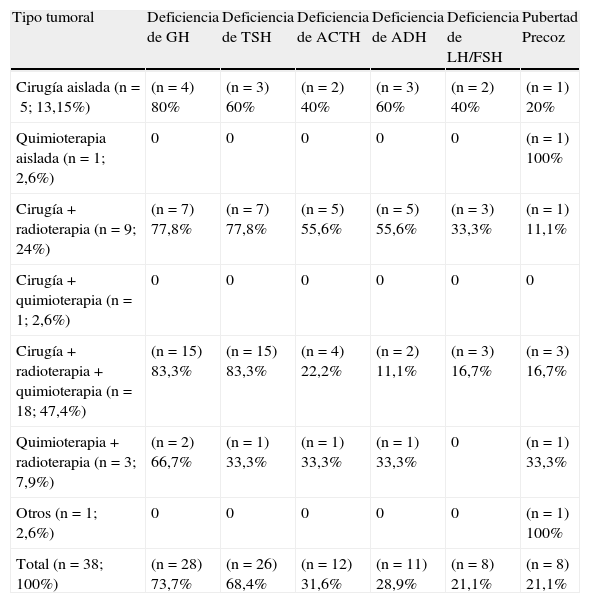
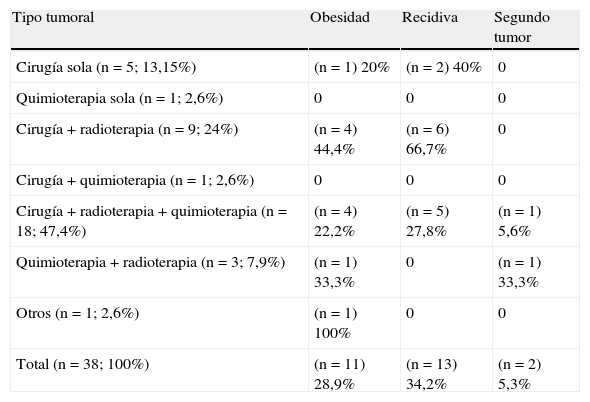
El tratamiento más frecuentemente administrado fue la combinación de cirugía+quimioterapia+radioterapia, empleado en el 47,4% de los pacientes; seguido de cirugía+radioterapia en un 24% de los casos. Los diferentes tipos de terapias empleadas y secuelas endocrinas están reflejados en las tablas 4 y 5. Ningún paciente recibió exclusivamente radioterapia. De los 30 pacientes que recibieron radioterapia (78,9% del total), en el 28,9% (n=11) fue en forma de irradiación holocraneal+espinal y en el 50% (n=19) exclusivamente irradiación craneal. Solo un paciente recibió quimioterapia aislada, tratándose de un varón con astrocitoma que comenzó con pubertad precoz a los 5,55 años de edad y que no ha manifestado ninguna alteración endocrina en su seguimiento, talla final +0,93 DE, sin recidivas ni segundas neoplasias. Un paciente diagnosticado de hamartoma hipotalámico a los 7,75 años recibió tratamiento con análogos de GnRH presentando evolutivamente obesidad a los 5 años de seguimiento.
Tratamientos recibidos y secuelas endocrinológicas. Número de casos (entre paréntesis) y porcentaje
| Tipo tumoral | Deficiencia de GH | Deficiencia de TSH | Deficiencia de ACTH | Deficiencia de ADH | Deficiencia de LH/FSH | Pubertad Precoz |
| Cirugía aislada (n=5; 13,15%) | (n=4) 80% | (n=3) 60% | (n=2) 40% | (n=3) 60% | (n=2) 40% | (n=1) 20% |
| Quimioterapia aislada (n=1; 2,6%) | 0 | 0 | 0 | 0 | 0 | (n=1) 100% |
| Cirugía+radioterapia (n=9; 24%) | (n=7) 77,8% | (n=7) 77,8% | (n=5) 55,6% | (n=5) 55,6% | (n=3) 33,3% | (n=1) 11,1% |
| Cirugía+quimioterapia (n=1; 2,6%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Cirugía+radioterapia+quimioterapia (n=18; 47,4%) | (n=15) 83,3% | (n=15) 83,3% | (n=4) 22,2% | (n=2) 11,1% | (n=3) 16,7% | (n=3) 16,7% |
| Quimioterapia+radioterapia (n=3; 7,9%) | (n=2) 66,7% | (n=1) 33,3% | (n=1) 33,3% | (n=1) 33,3% | 0 | (n=1) 33,3% |
| Otros (n=1; 2,6%) | 0 | 0 | 0 | 0 | 0 | (n=1) 100% |
| Total (n=38; 100%) | (n=28) 73,7% | (n=26) 68,4% | (n=12) 31,6% | (n=11) 28,9% | (n=8) 21,1% | (n=8) 21,1% |
Estrategias terapéuticas y casos con obesidad, recidiva y segundos tumores. Número de casos (entre paréntesis) y porcentaje
| Tipo tumoral | Obesidad | Recidiva | Segundo tumor |
| Cirugía sola (n=5; 13,15%) | (n=1) 20% | (n=2) 40% | 0 |
| Quimioterapia sola (n=1; 2,6%) | 0 | 0 | 0 |
| Cirugía+radioterapia (n=9; 24%) | (n=4) 44,4% | (n=6) 66,7% | 0 |
| Cirugía+quimioterapia (n=1; 2,6%) | 0 | 0 | 0 |
| Cirugía+radioterapia+quimioterapia (n=18; 47,4%) | (n=4) 22,2% | (n=5) 27,8% | (n=1) 5,6% |
| Quimioterapia+radioterapia (n=3; 7,9%) | (n=1) 33,3% | 0 | (n=1) 33,3% |
| Otros (n=1; 2,6%) | (n=1) 100% | 0 | 0 |
| Total (n=38; 100%) | (n=11) 28,9% | (n=13) 34,2% | (n=2) 5,3% |
IMC: índice de masa corporal.
El dato de talla final estaba disponible para 20 pacientes siendo de media −1,2±1,6. Ello corresponde a una disminución media de −0,53 DE respecto de su talla diana.
DiscusiónEste estudio muestra que tras 5 años de seguimiento de TSNC, un 76,3% de los casos presentaron una o más alteraciones endocrinológicas, dato similar a otras series (80%)12, si bien otros grupos registran tasas inferiores, del 501 o del 43%8. Los efectos neuroendocrinos pueden ser causados por daño hipotalámico (déficit de GH) o sobre órganos específicos (tiroides, gónadas)3.
En este estudio, el tipo tumoral más frecuente fue el meduloblastoma (26,3%), que, de acuerdo con la literatura, es uno de los tipos histológicos de TSNC más prevalentes. La histología del tumor per se no es indicador de la aparición de efectos a largo plazo, pero al variar el tratamiento según el tipo tumoral, la histología desempeña un papel indirecto indicando las secuelas3. La asociación cirugía+radioterapia+quimioterapia fue la terapia más empleada (47,4%). Actualmente, un 25% de los pacientes reciben neurocirugía solamente, un 40% cirugía+radioterapia y un 30% cirugía+radioterapia+quimioterapia, dependiendo de su edad, tipo histológico y localización tumoral1,8. Se asume que son pocos los efectos a largo plazo en aquellos tratados solo con cirugía8; esto contrasta con los datos de nuestro estudio donde estos pacientes cuentan con porcentajes altos de casi todas las alteraciones endocrinas. Se cree que los riesgos son elevados para aquellos tratados con radioterapia y cirugía, y todavía más elevados para aquellos que además reciben quimioterapia adyuvante8; como también se aprecia en nuestro estudio.
Existe una correlación fuerte entre la disfunción hipotálamo-hipofisaria y la radioterapia, que es dependiente de dosis13-15 y tiempo16,17. La edad a la que se recibe la radiación influye en algunas deficiencias hormonales1 (GH) pero no en otras18. Para el TSNC, la región hipotálamo-hipofisaria recibe una dosis media de 53,6Gy (40-70Gy). Tras irradiación craneal, se ha propuesto la siguiente secuencia de aparición de endocrinopatías19: GH (4,5 años), LHFSH (8 años), ACTH (9-10 años) y TSH (> 10 años)18. Hiperprolactinemia y diabetes insípida suelen acontecer después. La quimioterapia potencia los efectos deletéreos de la radioterapia sobre la función hipofisaria, pero no hay evidencia de que por sí sola produzca disfunción neuroendocrina17,20.
Eje de GH. El déficit de GH es la endocrinopatía más frecuente asociada a los TSNC en este y otros estudios3. Encontramos que el 73,7% tenía evidencia de déficit de GH, similar a otros estudios (70%6,13,14,21 o incluso 97%12), aunque en otras series fue de tan solo un 35,1%1 o un 21% de los casos8. Algunos de estos estudios han podido infraestimar las tasas de endocrinopatía al basar el diagnóstico en el autoinforme del paciente/familiares8, a diferencia de emplear resultados de laboratorio como en nuestro estudio y otros12. Recibir tratamiento a una edad más temprana se asocia con mayor prevalencia de deficiencia de GH3.
Tras la radioterapia craneal la deficiencia de GH es la complicación más frecuente, precoz6 y mejor estudiada, apareciendo en el 52-100% de los pacientes13. Con dosis entre 18-24Gy se suele detectar disfunción neurosecretora de GH, mientras que el déficit total de GH aparece con dosis > 30Gy (y aparece antes cuanto mayor dosis de radiación). Como en nuestro estudio, el riesgo de déficit de GH es elevado en meduloblastomas y gliomas ópticos, ya que las dosis de radioterapia son altas. El déficit de GH es irreversible y puede ocurrir desde 3 meses hasta 6 años después de la radioterapia14; siendo la media ± DE de años en nuestro estudio de 4,1±2,1. Esta disfunción es hipotalámica, por alteración de los mecanismos de contrarregulación gobernados por el sistema GHRH/somatostatina, dado que es más radiosensible que la hipófisis; dañándose esta última solo con dosis más altas de radiación22. Se acepta que es suficiente un test de estimulación de GH fallido tras radiación hipotálamo-hipofisaria para diagnosticar de déficit de GH23. Las publicaciones emplean diferentes pruebas (estimulación o secreción espontánea) con distinta sensibilidad para su diagnóstico, pudiendo haberse subestimado su incidencia según el test empleado18. En algunos pacientes con déficit de GH, los niveles de IGF-1 e IGFBP-3 pueden ser normales debido al sobrepeso y la regulación nutricional24.
La irradiación sobre el eje espinal retrasa el crecimiento de los cartílagos de crecimiento vertebrales y médula espinal, conduciendo a un tronco corto para unas desproporcionadamente largas extremidades (talla sentado más afectada que talla de pie)19,25. El meduloblastoma y el ependimoma reciben este tratamiento y llegan a perder hasta 9-10cm si la radiación es en el primer año de vida y 7cm si es a los 5 años.
No está claro el efecto de la quimioterapia sobre el cartílago de crecimiento20,26,27. Un estudio demostró que el efecto de la quimioterapia sobre la talla final era tan importante como la radioterapia espinal en los supervivientes a TSNC tratados con GH26. Los citotóxicos podrían amplificar el daño hipotálamo-hipofisario causado por la radiación17 y afectar a la producción hepática de IGF-124 y/o impedir su acción sobre la placa de crecimiento.
La talla final en nuestros pacientes queda dentro de la normalidad y presenta una diferencia media con la talla diana de −0,53 DE, mientras que otros estudios han publicado tallas finales significativamente más bajas que su talla media parental1, incluso con un 40% de las tallas finales por debajo del percentil 1026. La talla final inferior a la talla diana podría deberse a: déficit de GH, daño de la radioterapia sobre columna vertebral y los huesos largos, malnutrición por disminución de la ingesta, recidiva tumoral, quimioterapia, corticoterapia, pubertad precoz y otras endocrinopatías18. Existe asociación entre talla baja adulta y una menor edad al diagnóstico26,28.
Eje de TSH. Nuestra serie presenta una tasa elevada de hipotiroidismo (68,4%) frente a datos del 28% de otras cohortes15. Como consecuencia de la dosis de radioterapia craneal > 30Gy16,27,29, puede aparecer hipotiroidismo central, que es menos frecuente que el hipotiroidismo primario secundario a quimioterapia o radioterapia cervical15. Un estudio muestra tasas de hipotiroidismo central de un 6%30 y otro de hasta 14,9%1. Algunos estudios28,31 muestran una incidencia mayor de hipotiroidismo primario en aquellos supervivientes que han recibido radioterapia+quimioterapia (70-75%) que los que recibieron radioterapia exclusivamente (20-25%)31; esta asociación no ha sido encontrada en otros estudios32. Existe controversia respecto a si la radiación craneal exclusiva tiene menor riesgo de hipotiroidismo31 que la craneoespinal15. Se debate acerca de si para diagnosticar hipotiroidismo son suficientemente sensibles TSH y T4L o si sería necesario realizar el test de TRH o medir el ascenso de TSH nocturno18.
Eje gonadal. La pubertad precoz es típica de hamartomas, gliomas ópticos, ependimomas y astrocitomas. Si la dosis de radioterapia es > 18Gy pueden producir pubertad precoz/acelerada al interrumpir las influencias inhibitorias corticales sobre las neuronas GnRH hipotálamicas11. Paradójicamente, dosis > 30-50Gy18 también se asocian con hipogonadismo hipogonadotrófico11,15. Nuestro estudio y otros muestran cómo, a diferencia del déficit de GH y de TSH, que suelen manifestarse 2-3 años postratamiento, el déficit de GnRH no será manifiesto hasta la pubertad o edad adulta, haciendo su incidencia más difícil de cuantificar12. Apreciamos que un 21,1% de casos, similar a lo publicado (16-40%33) presentó progresión puberal escasa, retardada o amenorrea secundaria. Esto es más habitual encontrarlo en craneofaringiomas, histiocitosis de Langerhans y germinomas. No está claro si la quimioterapia aislada puede alterar la secreción de gonadotropinas.
Radioterapia y quimioterapia pueden producir afectación gonadal primaria6. Un estudio halló que la radiación espinal para TSNC se asociaba a un 35% de disfunción ovárica y un 3% de disfunción testicular12 (la radiación dispersada alcanza más, por proximidad, a los ovarios)4. Algunos quimioterápicos pueden producir déficits de hormonas sexuales en ambos géneros34. Estos efectos se pueden traducir en una fertilidad reducida34.
Eje de ACTH. Encontramos un 31,6% de déficit de ACTH en los supervivientes a TSNC, superior al 3-7,9%1 descrito en la literatura. Lo más habitual es encontrar supresión transitoria de ACTH debido al uso prolongado de corticoides en el tratamiento oncológico. También puede deberse al daño directo tumoral o quirúrgico sobre hipotálamo/hipófisis o a dosis de radioterapia > 30-50Gy27. Los estudios muestran una incidencia de deficiencia de ACTH del 19-38% tras radioterapia por TSNC, con una relación dosis-efecto16. La prueba de referencia para diagnosticar el déficit de ACTH sería el test de tolerancia insulínica, aunque la mayoría de los centros emplean el test corto de ACTH o la estimulación con glucagón; de ahí que la tasa de diagnóstico de esta deficiencia oscile18.
Otras endocrinopatías. Para que la diabetes insípida se manifieste clínicamente el déficit de ADH debe ser ≥ 90%. Fue diagnosticado en un 28,9% de nuestros pacientes, frente al 10,5%1 de otras series. Aparece más frecuentemente en aquellos tumores que afectan al infundíbulo (craneofaringioma, germinoma y glioma óptico)1, así como tras la cirugía. Aunque infrecuente, también se ha descrito con dosis de radioterapia > 45Gy35.
Dependiendo del área y cantidad de hipotálamo afectado aparecerán diferentes trastornos alimentarios, desde la caquexia diencefálica en los gliomas hipotalámicos, a la obesidad grave en los craneofaringiomas con afectación hipotalámica. Encontramos obesidad en un 28,9% de los pacientes, sobre todo tras la combinación cirugía+radioterapia, y a expensas de los craneofaringiomas. Es una cifra superior al 7% de otras series1 o incluso a otro estudio que halló que el IMC no difería de los controles emparejados por edad36. Otro estudio halló que dosis de radioterapia craneal > 20-50Gy, especialmente en niñas irradiadas a una edad menor a 4 años, asociaban significativamente obesidad hipotalámica10. Para los tumores que surgen y/o reciben tratamiento sobre el área selar, la obesidad es de tipo hipotalámica (40-50%), debida al daño al área ventromedial que es donde se integra la información de las hormonas periféricas (leptina, ghrelina e insulina). Esta disrupción conduciría a hiperfagia. Otros factores que contribuyen a esta obesidad serían: glucocorticoides, déficit de GH (aumento de masa grasa, disminución de fuerza) y menor actividad física10. Otras hipótesis serían: hiperestimulación vagal del hipotálamo sobre las células β-pancreáticas conduciendo a hiperinsulinsimo y obesidad, menor secreción de ghrelina, supresión posprandial de ghrelina reducida y el posible papel de melanocyte-stimulating hormone-α36.
Los segundos tumores debidos al tratamiento de TSNC son infrecuentes, pero de consecuencias potencialmente devastadoras. Surgen frecuentemente en el SNC11 (meningioma, astrocitoma maligno) pero también fuera de él (sarcoma de partes blandas, carcinoma tiroideo, carcinoma cutáneo y linfoma)37. El riesgo descrito de segundos tumores es del 1-2% a los 2-8 años21 o de 2,1% a los 20 años tras el tratamiento del TSNC38. Un estudio muestra que aquellos pacientes que recibieron radioterapia craneal ≥ 50Gy para TSNC tuvieron una incidencia de segunda neoplasia craneal del 7,1% a los 25 años del diagnóstico, comparado con un 1% en aquellos pacientes que no recibieron radioterapia11. La radiación craneal y/o espinal es un factor de riesgo para desarrollar nódulos tiroideos, siendo un alto porcentaje de ellos malignos18,35.
En adultos supervivientes a TSNC infantiles, la mortalidad a los 30 años del diagnóstico es del 25,8% (13 veces superior a controles emparejados por edad y sexo en EE. UU.) debido sobre todo a la recurrencia y/o progresión de la enfermedad primaria11. En nuestro estudio la recurrencia fue del 34,2%, debido en gran medida a la recidiva de craneofaringiomas.
En conclusión, el tipo de tumor y el tratamiento recibido determinará las secuelas endocrinas. Las deficiencias hormonales más frecuentes después de tratamiento de todos los tipos de TSNC, independientemente del tratamiento recibido, son GH y TSH. El diagnóstico precoz y una intervención temprana de la disfunción endocrina, reducen la morbilidad y pueden mejorar la calidad de vida a largo plazo. Un seguimiento médico de por vida es necesario ya que las complicaciones pueden aparecer años tras el tratamiento.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Presentación previa del trabajo: ciertos aspectos de este trabajo fueron presentados en forma de póster en la 51.ª Reunión Anual de la Sociedad Europea de Endocrinología Pediátrica (ESPE), celebrada en Leipzig (Alemania) entre los días 20 y 23 de septiembre del 2012. Los autores y título de la comunicación son: Güemes M, Fuente L, Caballero FJ, Martos-Moreno G, Muñoz-Calvo MT, Argente J. «Endocrinological effects in survivors of central nervous system tumors after a 5 year follow-up».
