

El síndrome de deleción 22q11.2 es un síndrome de genes contiguos con una incidencia de un caso por cada 4.000-6.000 recién nacidos. Posee una amplia variabilidad clínica y sus características clínicas más frecuentes son cardiopatía conotruncal, anomalías palatinas, hipocalcemia, problemas de inmunidad y de aprendizaje, y un fenotipo facial característico. El objetivo de este estudio es revisar las formas de presentación y las manifestaciones clínicas de los niños con deleción 22q11.2 como guía para su diagnóstico precoz.
Pacientes y métodosEstudio retrospectivo de 22 casos de deleción 22q11.2 diagnosticados en nuestro hospital entre los años 2004 y 2007, en que se analizan las siguientes variables: incidencia, sexo, edad en el momento del diagnóstico, forma de presentación, características clínicas, antecedentes familiares, mortalidad y evolución.
ResultadosDe los 22 pacientes, el 63% fueron varones y la edad media en el momento de realizar el diagnóstico fue de 4,5 años. Las formas de presentación fueron cardiopatía, retraso psicomotor, insuficiencia velopalatina, hipocalcemia y retraso mental o alteraciones psiquiátricas. Las principales manifestaciones clínicas fueron cardiopatía (84%), insuficiencia velopalatina (47%), retraso psicomotor y problemas de aprendizaje (79%). Todos los casos fueron deleciones de novo, salvo un caso en el que se identificó la deleción “en mosaico” en el padre. Fallecieron 3 pacientes a causa de cardiopatía.
ConclusionesLa expresión clínica es muy variable, aunque existe un fenotipo característico. Los niños con cardiopatía conotruncal son diagnosticados más tempranamente, pero en otras formas de presentación, como la disfagia congénita, el diagnóstico se retrasa más. Es necesario tener en cuenta las formas de presentación menos habituales para identificar en edades tempranas a estos pacientes y proporcionarles una atención multidisciplinaria temprana y un asesoramiento genético familiar adecuado.
The 22q11.2 deletion syndrome is a contiguous gene deletion syndrome with an incidence rate of 1/4,000-6,000 live births. The most specific clinical features are: congenital conotruncal heart diseases, palate anomalies, hypocalcaemia, immunity and learning problems, and a characteristic facial phenotype. The objective of this work is to review the presenting phenotype and clinical features of children with 22q11.2 deletion syndrome as a guide for early diagnosis.
Patients and methodsRetrospective study of 22 patients with 22q11.2 deletion syndrome diagnosed at our hospital in the time period 2004-2007. Variables analyzed: incidence, sex, age at diagnosis, presenting phenotype, clinical features, positive family history, mortality and natural history.
ResultsFrom a total of 22 patients, 63% were males, and the median age at diagnosis was of 4.5 years. Presenting phenotype: congenital heart disease, milestones delay, velopharyngeal incompetence, hypocalcaemia, and mental retardation/psychiatric disturbances. Clinical features:congenital heart disease (84%), velopharyngeal incompetence (47%), milestones delay and learning disabilities (79%). All of the deletions were de novo, except in one case where the deletion was present as mosaicism in the father. Three patients died, due to congenital heart disease.
ConclusionsClinical expression is widely variable, although a characteristic phenotype exists. Patients with heart disease are diagnosed earlier than other patients with unusual presenting phenotype such as congenital dysphagia. It is important to recognize less common phenotypes at early ages in order to provide multidisciplinary monitoring and accurate genetic counselling.
El síndrome de deleción 22q11.2 es un síndrome de genes contiguos causado por una pérdida de material genético en el brazo largo del cromosoma 22. Tiene una incidencia de un caso por cada 4.000-6.000 recién nacidos y no existen diferencias en cuanto a etnia o sexo1,2. Engloba una serie de síndromes descritos previamente en los que se ha identificado esta etiología común, como son los síndromes de DiGeorge, velocardiofacial o de Shprintzen, la anomalía facioconotruncal y el síndrome cardiofacial de Cayler3–7. Inicialmente se agruparon todas estas entidades bajo el epónimo CATCH 22, pero hoy día se prefiere la denominación de síndrome de deleción 22q11.23.
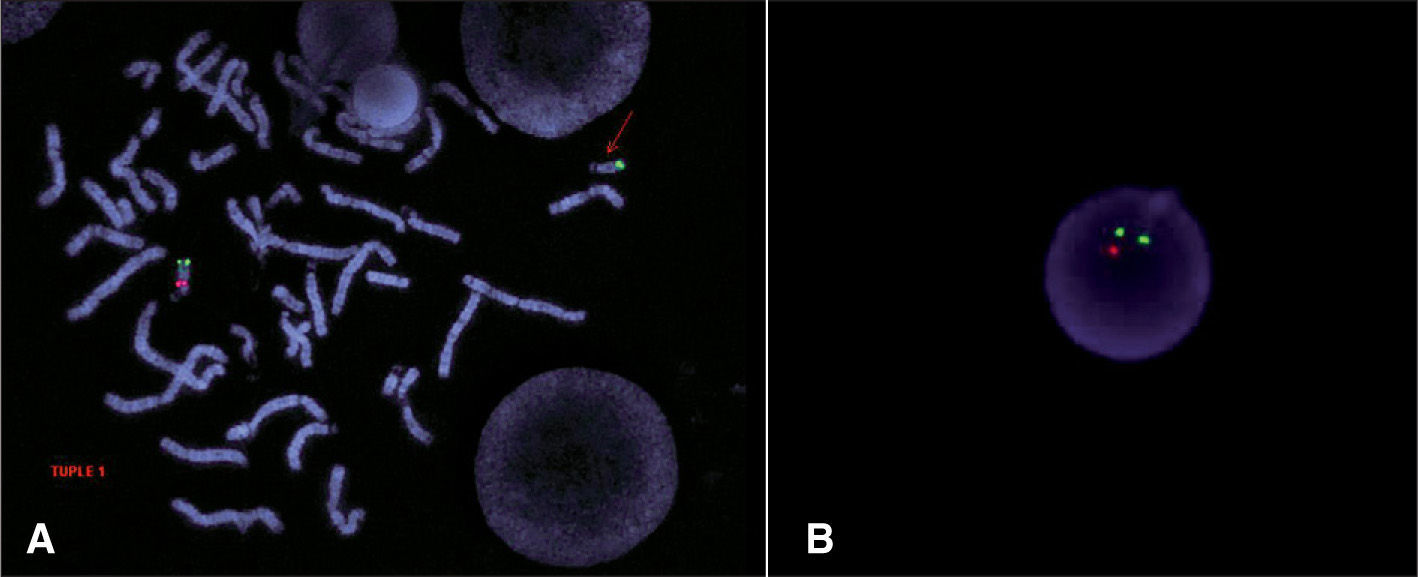
El diagnóstico citomolecular se realiza mediante hibridación in situ fluorescente (FISH), utilizando una sonda de ADN de la región crítica de DiGeorge en 22q, que abarca unas 5 Mb8,9 (fig. 1). Mediante esta técnica se identifica un 95 % de los casos y pueden descartarse posibles reorganizaciones cromosómicas que abarquen esta región como, por ejemplo, translocaciones (hecho que ocurre en menos del 1 % de los casos)2. Se ha descrito una deleción frecuente de 3Mb y una de menor tamaño (1,5Mb) y no parece existir relación entre el tamaño de la deleción y la gravedad de la expresividad clínica2,8,9.
 imagen de FISH en la que se observa la ausencia de señal roja en uno de los cromosomas 22. Sonda roja: sonda de la región crítica. Sonda verde: sonda control. B) imagen de FISH en interfase en la que se observa la ausencia de señal roja en uno de los cromosomas 22.")
Deleción 22q11.2. A) imagen de FISH en la que se observa la ausencia de señal roja en uno de los cromosomas 22. Sonda roja: sonda de la región crítica. Sonda verde: sonda control. B) imagen de FISH en interfase en la que se observa la ausencia de señal roja en uno de los cromosomas 22.
Este síndrome tiene un espectro fenotípico muy amplio, por lo que es importante establecer signos guía de diagnóstico clínico10–12. La cardiopatía es la anomalía más frecuente (en el 74 % de los pacientes), y las malformaciones conotruncales son las más habituales (tetralogía de Fallot, comunicación interventricular, truncus arteriosus y coartación aórtica). Las anomalías palatinas (incompetencia velofaríngea y paladar hendido submucoso) aparecen en casi el 70 % de los casos. También son características la hipocalcemia por hipoparatiroidismo funcional y las alteraciones inmunitarias que ocasionan infecciones de repetición, sobre todo en el área sinopulmonar. Entre el 70 y el 90 % de los casos tienen dificultades para el aprendizaje y el 46 % presenta retraso mental. En la adolescencia o en la vida adulta se han descrito trastornos psiquiátricos del tipo de psicosis y alteraciones del estado de ánimo11,13. Estos niños presentan una facies característica y reconocible.
Los pacientes son atendidos por pediatras, cardiólogos infantiles, otorrinolaringólogos, neuropediatras o psiquiatras. En ocasiones, el diagnóstico se retrasa debido a su expresividad variable y a las formas atípicas de presentación.
El objetivo del trabajo es revisar las características clínicas y la evolución de nuestros pacientes con microdeleción 22q11.2, para un mejor conocimiento de su historia natural y formas de presentación, que favorezcan el diagnóstico precoz del síndrome.
PACIENTES Y MÉTODOSSe ha realizado una revisión de 22 pacientes con deleción 22q11.2 evaluados clínicamente en la unidad de genética médica de nuestro hospital. El estudio citogenético se realizó a los niños y a sus padres en el servicio de citogenética del hospital mediante cariotipo convencional y citogenética molecular (FISH) de la región 22q11.2 con las sondas comerciales N25 (ONCOR) y TUPLE 1 (Vysis). Se analizaron las siguientes variables: incidencia, sexo, edad en el momento del diagnóstico, forma de presentación, frecuencia de anomalías asociadas, origen de la deleción, evolución y mortalidad.
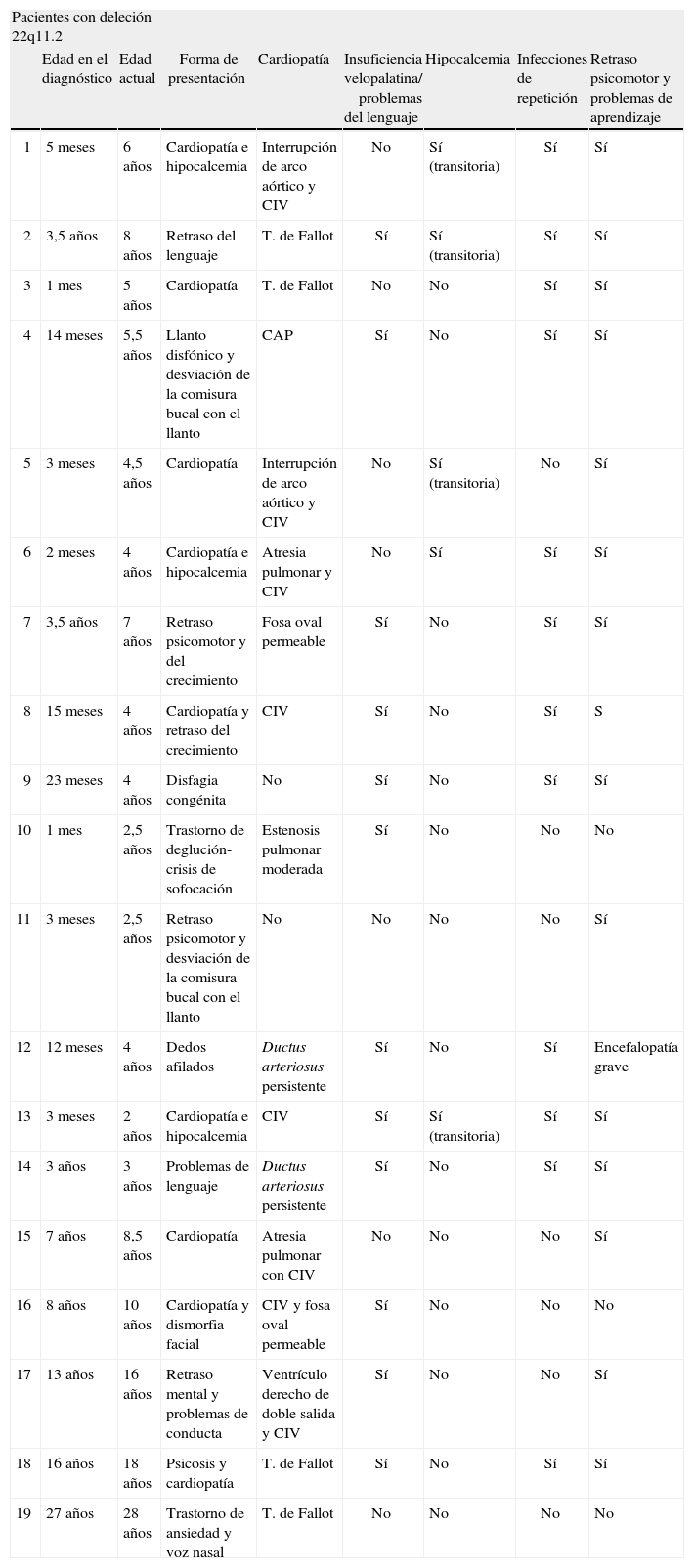
RESULTADOSNúmero de pacientes, sexo y mortalidadSe han revisado 22 pacientes, de los que un 63% son varones (14 varones y 8 mujeres). Tres pacientes fallecieron en el período de lactante por complicaciones tras la cirugía cardíaca. Se observan dos grupos distintos de edad debido a la recuperación de pacientes que han sido diagnosticados en la adolescencia y en la edad adulta. Un grupo comprende a pacientes desde el nacimiento hasta los 7 años de edad y otro grupo menor es el de 5 pacientes con edades comprendidas entre los 8,5 y 28 años (tabla 1).
Resumen de las manifestaciones clínicas de los pacientes con deleción 22q11.2
| Pacientes con deleción 22q11.2 | ||||||||
| Edad en el diagnóstico | Edad actual | Forma de presentación | Cardiopatía | Insuficiencia velopalatina/ problemas del lenguaje | Hipocalcemia | Infecciones de repetición | Retraso psicomotor y problemas de aprendizaje | |
| 1 | 5 meses | 6 años | Cardiopatía e hipocalcemia | Interrupción de arco aórtico y CIV | No | Sí (transitoria) | Sí | Sí |
| 2 | 3,5 años | 8 años | Retraso del lenguaje | T. de Fallot | Sí | Sí (transitoria) | Sí | Sí |
| 3 | 1 mes | 5 años | Cardiopatía | T. de Fallot | No | No | Sí | Sí |
| 4 | 14 meses | 5,5 años | Llanto disfónico y desviación de la comisura bucal con el llanto | CAP | Sí | No | Sí | Sí |
| 5 | 3 meses | 4,5 años | Cardiopatía | Interrupción de arco aórtico y CIV | No | Sí (transitoria) | No | Sí |
| 6 | 2 meses | 4 años | Cardiopatía e hipocalcemia | Atresia pulmonar y CIV | No | Sí | Sí | Sí |
| 7 | 3,5 años | 7 años | Retraso psicomotor y del crecimiento | Fosa oval permeable | Sí | No | Sí | Sí |
| 8 | 15 meses | 4 años | Cardiopatía y retraso del crecimiento | CIV | Sí | No | Sí | S |
| 9 | 23 meses | 4 años | Disfagia congénita | No | Sí | No | Sí | Sí |
| 10 | 1 mes | 2,5 años | Trastorno de deglución-crisis de sofocación | Estenosis pulmonar moderada | Sí | No | No | No |
| 11 | 3 meses | 2,5 años | Retraso psicomotor y desviación de la comisura bucal con el llanto | No | No | No | No | Sí |
| 12 | 12 meses | 4 años | Dedos afilados | Ductus arteriosus persistente | Sí | No | Sí | Encefalopatía grave |
| 13 | 3 meses | 2 años | Cardiopatía e hipocalcemia | CIV | Sí | Sí (transitoria) | Sí | Sí |
| 14 | 3 años | 3 años | Problemas de lenguaje | Ductus arteriosus persistente | Sí | No | Sí | Sí |
| 15 | 7 años | 8,5 años | Cardiopatía | Atresia pulmonar con CIV | No | No | No | Sí |
| 16 | 8 años | 10 años | Cardiopatía y dismorfia facial | CIV y fosa oval permeable | Sí | No | No | No |
| 17 | 13 años | 16 años | Retraso mental y problemas de conducta | Ventrículo derecho de doble salida y CIV | Sí | No | No | Sí |
| 18 | 16 años | 18 años | Psicosis y cardiopatía | T. de Fallot | Sí | No | Sí | Sí |
| 19 | 27 años | 28 años | Trastorno de ansiedad y voz nasal | T. de Fallot | No | No | No | No |
CAP: conducto arterial permeable; CIV: comunicación interventricular; T. de Fallot: tetralogía de Fallot.
Se ha calculado una incidencia en nuestro medio de un caso por cada 6.800 recién nacidos vivos basado en el grupo de menor edad. Los pacientes fueron remitidos sobe todo desde los servicios de cardiología infantil (8 casos), neuropediatría (7 casos), pediatría general (2 casos) y psiquiatría (3 casos). La edad media en el momento del diagnóstico fue de 14 meses si se considera sólo el grupo de menor edad y de 4,5 años si se incluye el grupo de pacientes diagnosticados a edades más avanzadas.
Origen de la deleciónTras realizar el estudio citomolecular (cariotipo y FISH de la región 22q11.2) tanto en el paciente como en sus padres, se ha observado que 21 casos eran deleciones de novo; en un caso se identificó la deleción "en mosaico" en el padre cuyo fenotipo facial era indicativo.
Formas de presentaciónLa forma de presentación más frecuente fue la cardiopatía (41 %), seguida de retraso psicomotor (21 %), problemas de deglución o insuficiencia velopalatina (16 %), e hipocalcemia (16%). El retraso mental ± alteraciones psiquiátricas fueron la forma de presentación en un 10% de los casos si se considera a todos los pacientes, siendo del 40 % en el grupo de mayor edad. Dos de las pacientes iniciaron la enfermedad con disfagia congénita aislada y precisaron la colocación de una sonda de gastrostomía para su alimentación. En una de ellas se detectó posteriormente una estenosis valvular pulmonar moderada. El fenotipo facial se hizo evidente con la evolución de estas pacientes y la disfagia fue mejorando a partir de los 2 años de edad, consiguiendo alimentarse por completo por vía oral.
Cuadro clínicoEn la tabla 1 se exponen las manifestaciones clínicas de los pacientes. No se incluyen los 3 pacientes que fallecieron precozmente por su cardiopatía al no disponer de datos evolutivos.
CardiopatíaLa cardiopatía se identificó en un 89 % de los pacientes, y los defectos más frecuentes fueron tetralogía de Fallot (25%), atresia pulmonar con comunicación interventricular (CIV) (12,5%) y CIV aislada (19%). La edad media en el momento de realizar el diagnóstico, en los pacientes del grupo de menor edad, y con estos defectos cardíacos, fue de 10 meses, siendo de 21 meses en los casos que presentaban asociada otro tipo de cardiopatía.
Anomalías palatinas, laríngeas y alteraciones de la degluciónEl 47 % de los pacientes presentaron insuficiencia velopalatina y/o problemas de deglución con regurgitación nasal y succión débil. Tres pacientes precisaron la colocación de una sonda de gastrostomía para alimentarse y otro requirió alimentación por sonda nasogástrica hasta los 6 meses de edad. Sólo una paciente presentó paladar hendido submucoso. Un paciente tuvo anomalías laríngeas con estenosis subglótica congénita tipo 1, membrana laríngea y laringomalacia que precisó laringoplastia con injerto costal. El 21 % presentó problemas en la articulación del lenguaje, con problemas de hipernasalidad (21 %) y escape aéreo, siendo ésta la forma de presentación en dos de ellos.
HipocalcemiaLa hipocalcemia fue transitoria en el 21 % de los casos, y sólo en un paciente fue secundaria a hipoparatiroidismo primario.
Timo, infecciones y autoinmunidadEl 58 % de los pacientes presentó infecciones recurrentes, sobre todo en el área sinopulmonar (otitis media y neumonía), y la incidencia de hiperreactividad bronquial-asma fue del 10,5%. Se observó agenesia tímica en un paciente. En relación con el espectro inmunológico, el 53 % de los pacientes presentó un número disminuido de linfocitos T totales, con una alteración en la respuesta a mitógenos en el 21 %. En el 10 % se observaron inmunodeficiencia humoral o alteraciones en el complemento. No se detectó positividad de autoanticuerpos ni de marcadores de celiaquía en ninguno de los pacientes, y el estudio tiroideo fue normal en todos los casos. Coincidieron en un paciente dos fenómenos autoinmunes, la enfermedad de Kawasaki y la púrpura trombocitopénica idiopática crónica recidivante. Respecto a las vacunas, el 73 % de los pacientes recibió la vacunación completa sin que en ningún caso hubiera reacciones adversas.
Fenotipo facialAunque a veces es difícil identificarlo en el nacimiento, todos los pacientes presentaron evolutivamente rasgos faciales característicos (fig. 2). Tienen hendiduras palpebrales estrechas, raíz nasal prominente y cuadrada, con punta bulbosa y narinas pequeñas y antevertidas. Es frecuente la retromicrognatia. Un hallazgo característico es la desviación de la comisura bucal con el llanto. Los pabellones auriculares son prominentes, con un hélix muy marcado. Los dedos son largos y afilados.
Retraso psicomotor, problemas de aprendizaje y anomalías neurológicas
El 79 % de los pacientes presentaron retraso psicomotor y problemas en el aprendizaje y el 26 %, convulsiones o equivalentes convulsivos (crisis febriles en 2 pacientes y espasmos del sollozo en otro). Las convulsiones se asociaron con hipocalcemia en una paciente y en otro caso se produjeron en el contexto de una encefalopatía hipóxico-isquémica. Un paciente presentaba epilepsia (5%). En el 26% se observaron anomalías cerebrales estructurales: gliosis frontal derecha, alteración en la formación del lóbulo temporal medial y basal bilateral, retracción difusa parenquimatosa y megacisterna magna en 2 pacientes.
Otras anomalías asociadasEl 21% de los pacientes presentó microcefalia, el 15,7% talla baja y otro 15,7%, anomalías nefrourológicas (hidronefrosis y reflujo vesicoureteral). Se identificaron anomalías oculares en 3 pacientes, dacriocistitis en dos y estrabismo en uno.
DISCUSIÓNLa expresión clínica del síndrome de deleción 22q11.2 es extremadamente variable, lo que conlleva que en ocasiones su diagnóstico resulte difícil12. El mejor conocimiento de esta entidad por parte de los distintos especialistas médicos ha adelantado su diagnóstico en los últimos años, como se evidencia en los dos grupos de edad. La incidencia observada en nuestra población fue de un caso por cada 6.800 recién nacidos, en el grupo de menor edad, sin que se observaran diferencias en cuanto al sexo. La edad media en el momento del diagnóstico en este grupo fue de 14 meses, y los casos más tempranos fueron los que presentaban una cardiopatía típica (conotruncal, defectos del arco aórtico y CIV). El origen de la deleción fue de novo en el 95 % de los casos, dato similar a lo descrito en la bibliografía médica (93 %)2.
La demora en el diagnóstico en el grupo de menor edad se produjo en los casos con problemas de deglución, retraso psicomotor o de aprendizaje, que se hicieron más evidentes con el paso del tiempo. El grupo de pacientes diagnosticados en la adolescencia, a pesar de tener un fenotipo clásico previo, fueron identificados al ser remitidos para llevar a cabo una evaluación por trastornos psiquiátricos y anomalías congénitas. El desconocimiento de este síndrome en el pasado hizo que estos pacientes pasaran desapercibidos en la infancia y fueran diagnosticados en edades más avanzadas por fenotipos característicos e identificables por personal sensibilizado11,13,14.
Es importante que los especialistas con un contacto más frecuente con estos pacientes como son cardiólogos, neurólogos, psiquiatras y, por supuesto, el pediatra general, se familiaricen con las distintas formas de presentación.
La forma más frecuente de presentación fue la cardiopatía (en el 42 % de los pacientes). Algún tipo de cardiopatía se identificó en el 89 % de los casos (frente al 75 % descrito en la bibliografía médica)2.
El retraso psicomotor y/o del aprendizaje fue la segunda forma más frecuente de presentación en niños con edades más avanzadas, lo que enfatiza la importancia de sospechar este diagnóstico en la presencia de retraso y valorar la existencia de otros síntomas indicativos. Destacamos la disfagia congénita como forma de presentación poco habitual, lo que retrasó el diagnóstico en dos de nuestras pacientes. Es preciso considerar el síndrome de deleción 22q11.2 en el diagnóstico diferencial de la disfagia congénita.
La insuficiencia velopalatina, así como los problemas de articulación del lenguaje (rinolalia abierta), son muy característicos15. Las alteraciones típicas en la pronunciación de estos pacientes son hipernasalidad, consonantes de baja presión y articulación compensatoria que se deben a la insuficiencia velofaríngea. Los problemas de deglución también son frecuentes, con atragantamientos y reflujo nasal del alimento. Como dato de buen pronóstico, estos problemas de deglución suelen ir mejorando con la edad, y en la mayoría de los casos se consigue la alimentación por vía oral hacia los 2–3 años, como se ha observado en nuestros casos.
La hipocalcemia fue un hallazgo poco frecuente. Dentro de las anomalías endocrinológicas también debe evaluarse la función tiroidea, que no estuvo alterada en ninguno de nuestros pacientes.
Se ha observado inmunodeficiencia hasta en un 77 % de los pacientes en otras series16–19. Suele ser una inmunodeficiencia celular leve-moderada de células T con una disminución en el número total de linfocitos T y una respuesta proliferativa pobre ante mitógenos. No suelen presentar infecciones oportunistas, ya que la inmunodeficiencia no es grave, pero sí suelen aparecer infecciones recurrentes en el área sinopulmonar. Las infecciones recurrentes fueron un hallazgo frecuente en nuestros pacientes (58 %), sobre todo las otitis medias y las neumonías. En ningún caso fue éste el signo guía de sospecha, aunque sí lo ha sido en otras series10. Sólo en un paciente se ha objetivado agenesia de timo con inmunodeficiencia celular y neumonías de repetición. En el estudio inmunológico realizado se detectó una inmunodeficiencia celular transitoria en edades tempranas (53%), que se normalizó con el tiempo y se observó una inmunodeficiencia humoral en un 10,5 %, que puede deberse a la evolución normal de la inmunidad en lactantes.
Se han planteado dudas sobre la posibilidad de que estos pacientes puedan seguir o no el calendario vacunal habitual. En nuestra serie no se han observado reacciones adversas a la vacunación en ninguno de los pacientes que siguieron una vacunación reglada, posiblemente debido a que las alteraciones inmunológicas son transitorias20. Estos pacientes tienen también una mayor probabilidad para desarrollar enfermedades autoinmunes, como púrpura trombocitopénica idiopática (PTI), hipotiroidismo o hipertiroidismo (enfermedad de Graves), enfermedad celíaca y anemia hemolítica, lo que debe tenerse en cuenta en el seguimiento. La artritis reumatoide juvenil es 150 veces más frecuente en estos niños que en la población general16. Sólo uno de los pacientes presentó fenómenos autoinmunes en nuestra serie (PTI y enfermedad de Kawasaki).
Respecto al fenotipo facial, a veces no es fácil identificar los rasgos faciales característicos, sobre todo en los primeros meses de vida. En nuestros casos observamos el fenotipo facial típico indicativo en todos los pacientes, que se hizo más evidente a medida que crecieron. Una característica frecuente fue la desviación de la comisura bucal con el llanto, que ayudó al diagnóstico en 3 pacientes. El padre de una paciente resultó presentar un mosaicismo para la deleción. Este mosaicismo se detectó al ampliar el estudio mediante FISH por alta sospecha clínica por el fenotipo facial (hendiduras palpebrales estrechas y raíz nasal cuadrada). Es importante, por tanto, estudiar un número suficiente de células en los individuos con sospecha clínica para descartar el mosaicismo.
La gran mayoría de nuestros pacientes (79 %) presentaron retraso psicomotor y problemas de aprendizaje, y fue ésta la forma de presentación en un 21 % de los casos, por lo que debe considerarse el diagnóstico en niños que acudan con este problema. Los pacientes de mayor edad se han identificado por retraso mental, problemas de conducta y ansiedad, características que sirven de guía, ya que suelen estar presentes en un alto porcentaje (90 %) en la adolescencia y en la vida adulta10. La epilepsia en nuestra serie tiene una frecuencia similar a la publicada (7 %)21.
Otras anomalías asociadas al síndrome son las nefrourológicas2, que se han detectado también en nuestros pacientes (hidronefrosis y reflujo vesicoureteral), así como las esqueléticas (polidactilia preaxial y postaxial, y malformaciones vertebrales y costales)2. Se ha descrito en la bibliografía médica un paciente con deleción 22q11.2 con una hemimelia22, así como una familia con malformación de la mano y pie hendido en la que se identificó una monosomía 22q11.2 y 17p13 323. Ninguno de nuestros pacientes presentó anomalías esqueléticas ni alteraciones de los miembros.
El estudio genético debe ampliarse a los padres para ofrecer un asesoramiento genético adecuado. Debe indicarse el estudio prenatal específico en caso de identificar, mediante ecografía, cardiopatía conotruncal y/o anomalías palatinas en el feto.
En resumen, el síndrome de deleción 22q11.2 debe plantearse en neonatos o lactantes con cardiopatía congénita conotruncal, anomalías palatinas y/o retraso psicomotor, que puedan presentar o no un fenotipo facial indicativo u otras de las anomalías asociadas como, por ejemplo, hipocalcemia. A partir del año debe considerarse este diagnóstico en presencia de retraso de aprendizaje o retraso mental y fenotipo conductual o características faciales indicativas. Se deben también tener en cuenta formas de presentación menos típicas, como la disfagia congénita.
Un diagnóstico precoz de este síndrome será muy útil para poder ofrecer a estos pacientes una atención multidisciplinaria adecuada y un correcto seguimiento clínico, actuando de forma precoz sobre las posibles anomalías asociadas.
AgradecimientosA todos los pacientes y a sus familias, que han permitido la publicación de estos datos, así como a los distintos especialistas que intervienen en la atención y el seguimiento multidisciplinario de estos pacientes.

