








La hiperplasia suprarrenal congénita debida al déficit de 21-hidroxilasa es una enfermedad autosómica recesiva causada por mutaciones en el gen CYP21A2. En las formas clásicas se produce defecto de cortisol y aldosterona (insuficiencia suprarrenal y pérdida salina) y virilizacion de la recién nacida afecta con ambigüedad genital. En este artículo ofrecemos algunas recomendaciones para el diagnóstico, que debe ser lo más precoz posible, y el tratamiento, adecuado e individualizado. El estudio genético del paciente y su familia es clave en el diagnóstico del propio afectado, y también permite establecer el consejo genético, así como el diagnóstico y tratamiento prenatales en futuros embarazos.
Congenital adrenal hyperplasia due to 21-hydroxylase deficiency is an autosomal recessive disorder caused by mutations in the CYP21A2 gene. Cortisol and aldosterone synthesis are impaired in the classic forms (adrenal insufficiency and salt-wasting crisis). Females affected are virilised at birth, and are at risk for genital ambiguity. In this article we give recommendations for an early as possible diagnosis and an appropriate and individualised treatment. A patient and family genetic study is essential for the diagnosis of the patient, and allows genetic counselling, as well as a prenatal diagnosis and treatment for future pregnancy.
La hiperplasia suprarrenal congénita (HSC) es una enfermedad autosómica recesiva producida por fallo en la esteroidogénesis suprarrenal. El 90-95% de casos presentan déficit del enzima 21-hidroxilasa (21OHD, OMIM#201910) con bloqueo variable en la síntesis de glucocorticoides y mineralocorticoides y producción excesiva de andrógenos originando diversos cuadros clínicos que pueden manifestarse en el periodo neonatal (formas «clásicas pierde sal» y «clásicas virilizantes simples») o durante la infancia, adolescencia o edad adulta (algunas «virilizantes simples» y formas «no clásicas»)1,2.
El tratamiento en las formas clásicas es complejo, individualizado, multidisciplinar y requiere la implantación de un programa estructurado de intervención y seguimiento, por lo que deberían atenderse en centros clínicos de referencia (CCR).
Diversas sociedades científicas (sociedades de endocrinología pediátrica europea, americana, australiana, japonesa) y asociaciones de pacientes han editado guías de actuación para esta enfermedad.
El Grupo de Trabajo sobre Hiperplasia Suprarrenal Congénita de la Sociedad Española de Endocrinología Pediátrica, basándonos en recomendaciones publicadas y en nuestra experiencia en formas clásicas, ofrece en este artículo recomendaciones generales que pueden ser útiles a pediatras que tratan a pacientes con 21OHD.
EpidemiologíaLa incidencia es variable según las poblaciones estudiadas y recogida de datos, extraídos o no de programas de detección precoz: para formas clásicas en recién nacidos es de 1/10.000-1/20.0002, siendo en nuestro medio de 1/10.000-1/14.0003, lo que implica 1/50-1/60 de portadores de mutación grave.
Fisiopatología y formas clínicasLa 21-hidroxilación suprarrenal se realiza por el enzima esteroide-21hidroxilasa (también denominado citocromo P450c21) que convierte progesterona en deoxicorticosterona y 17-hidroxiprogesterona (17OHP) en 11-deoxicortisol, culminando en la síntesis de aldosterona y cortisol, respectivamente. La gravedad del compromiso funcional del enzima determina la clínica (fig. 1).

21OHD provoca una disminución de la síntesis de hormonas situadas por debajo del bloqueo, glucocorticoides y mineralocorticoides, aumento de los productos previos como 17OHP y aumento de síntesis de la vía de los andrógenos. El déficit de cortisol conlleva aumento de corticotropina (ACTH) e hiperplasia de la glándula4.
En las formas graves existen otras vías metabólicas afectadas: disminución de producción de adrenalina en la médula adrenal (tendencia al colapso cardiovascular)5 y activación de síntesis de dihidrotestosterona (DHT) por vías metabólicas quiescentes aumentando la virilización del feto femenino6.
Las formas clínicas más graves se manifiestan en el periodo neonatal por el déficit de cortisol y aldosterona con pérdida salina potencialmente mortal1. El aumento de andrógenos origina virilización y ambigüedad de los genitales externos en las niñas recién nacidas. Es muy útil la clasificación de Prader para describir el grado de virilización (fig. 2). Posteriormente, sin el adecuado tratamiento, se produciría aceleración de la maduración ósea, cierre precoz de las epífisis, pseudopubertad precoz y talla baja adulta.
 Estadios de Prader. Prader 1: genitales externos femeninos; Prader 5: genitales externos masculinos. b) Recién nacida afecta de 21OHD. Es fácil en una primera valoración asignarle sexo masculino (Prader 5).")
Las formas menos graves comienzan en la infancia tardía, adolescencia o edad adulta; se caracterizan por signos de androgenización: pubarquia prematura, aceleración de la velocidad de crecimiento y maduración ósea, hirsutismo, acné severo y oligomenorrea, sin déficit evidente de aldosterona; pueden pasar desapercibidas, siendo elevado el número de diagnósticos tardíos.
Con frecuencia 21OHD provoca situaciones de gravedad intermedia.
Diagnóstico de déficit de 21-hidroxilasaSe sospechará 21OHD ante:
- •
Niño/a con clínica de pérdida salina en las primeras semanas de vida.
- •
Niñas virilizadas al nacimiento o inicio de virilización posnatal, pubarquia o pubertad precoz.
- •
Niños con inicio de virilización en la infancia.
El diagnóstico se basa en el análisis del esteroide previo al bloqueo: 17OHP.
Diagnóstico en el recién nacidoPrograma de detección precoz: cribado neonatalLa detección precoz de 21OHD, recomendada internacionalmente2, solo se realiza en algunas comunidades autónomas. Se basa en determinar 17OHP en muestra de sangre capilar el segundo día de vida, junto con la detección precoz de otras enfermedades. Sus objetivos son: anticiparse a la aparición de una crisis de pérdida salina, evitar la incorrecta asignación de sexo y diagnosticar precozmente las formas virilizantes simples7.
Cada laboratorio de referencia debe establecer los valores normales para su población según la edad gestacional, el peso al nacimiento y el sexo. Aunque actualmente se emplea fluorinmunoensayo (DELFIA), la espectrofotometría de masas permitirá excluir reacciones cruzadas de distintos esteroides suprarrenales. El genotipado CYP21A2 es útil como prueba de segundo nivel8.
La administración prenatal de glucocorticoides puede disminuir el nivel de 17OHP e induce falsos negativos. El uso de espironolactona puede producir un resultado positivo.
La tabla 1 muestra el algoritmo diagnóstico ante un resultado «positivo».
Algoritmo diagnóstico: recién nacido detectado por cribado neonatal (DELFIA) (nacido a término y con peso adecuado para su edad gestacional)
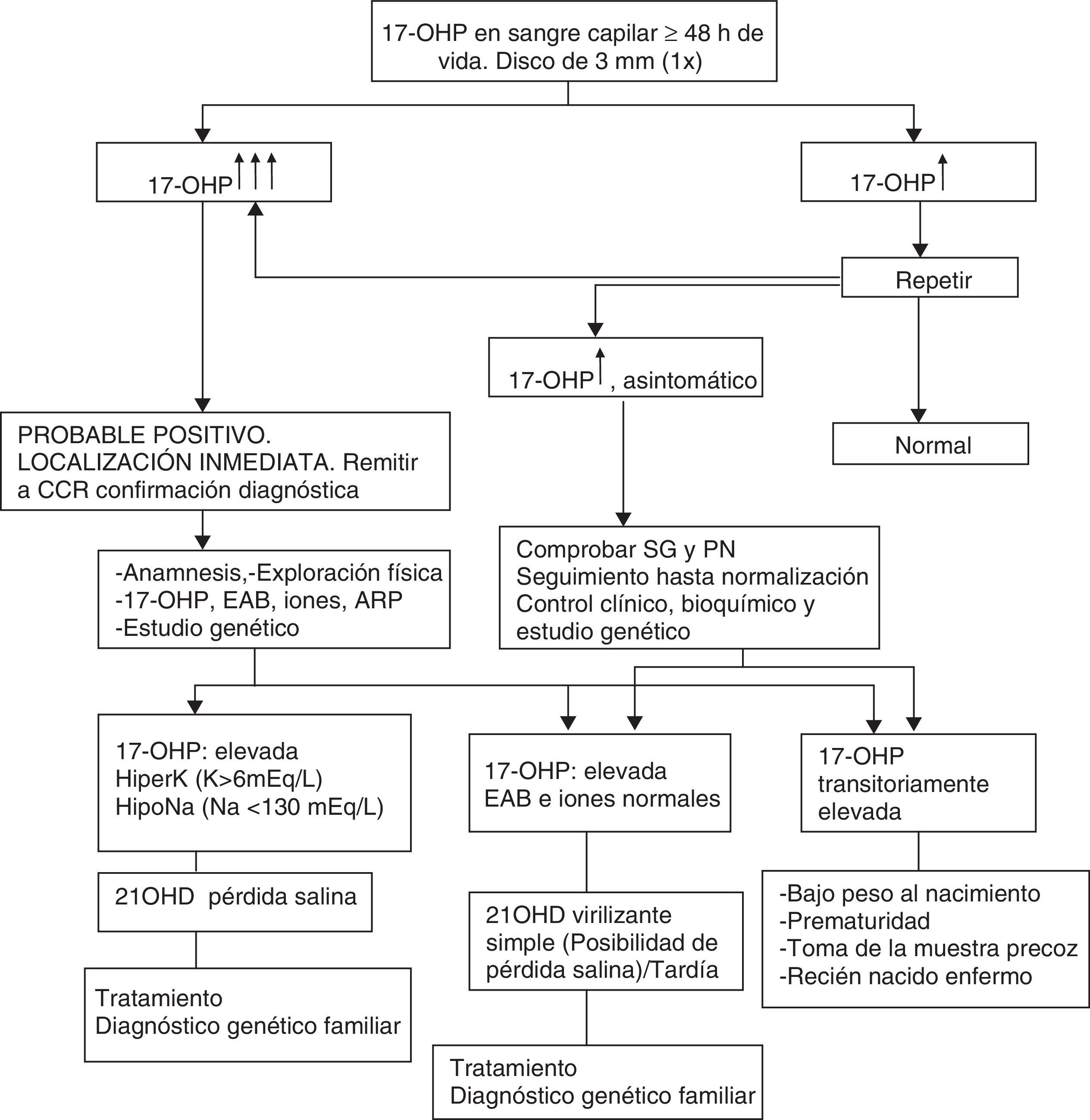
17OHP: 17-hidroxiprogesterona; 21OHD: deficiencia de 21-hidroxilasa; ARP: actividad de renina plasmática; CCR: centro clínico de referencia; EAB: equilibrio ácido-base; HiperK: Hiperpotasemia; HipoN: Hiponatremia; PN: peso al nacimiento; SG: semanas de gestación.
La tabla 2 recoge la actuación ante un recién nacido con sospecha de 21OHD, basado en la elevación de 17OHP excluyendo valores transitoriamente elevados (prematuridad, bajo peso, enfermedad neonatal grave) que suelen normalizarse antes del año8.
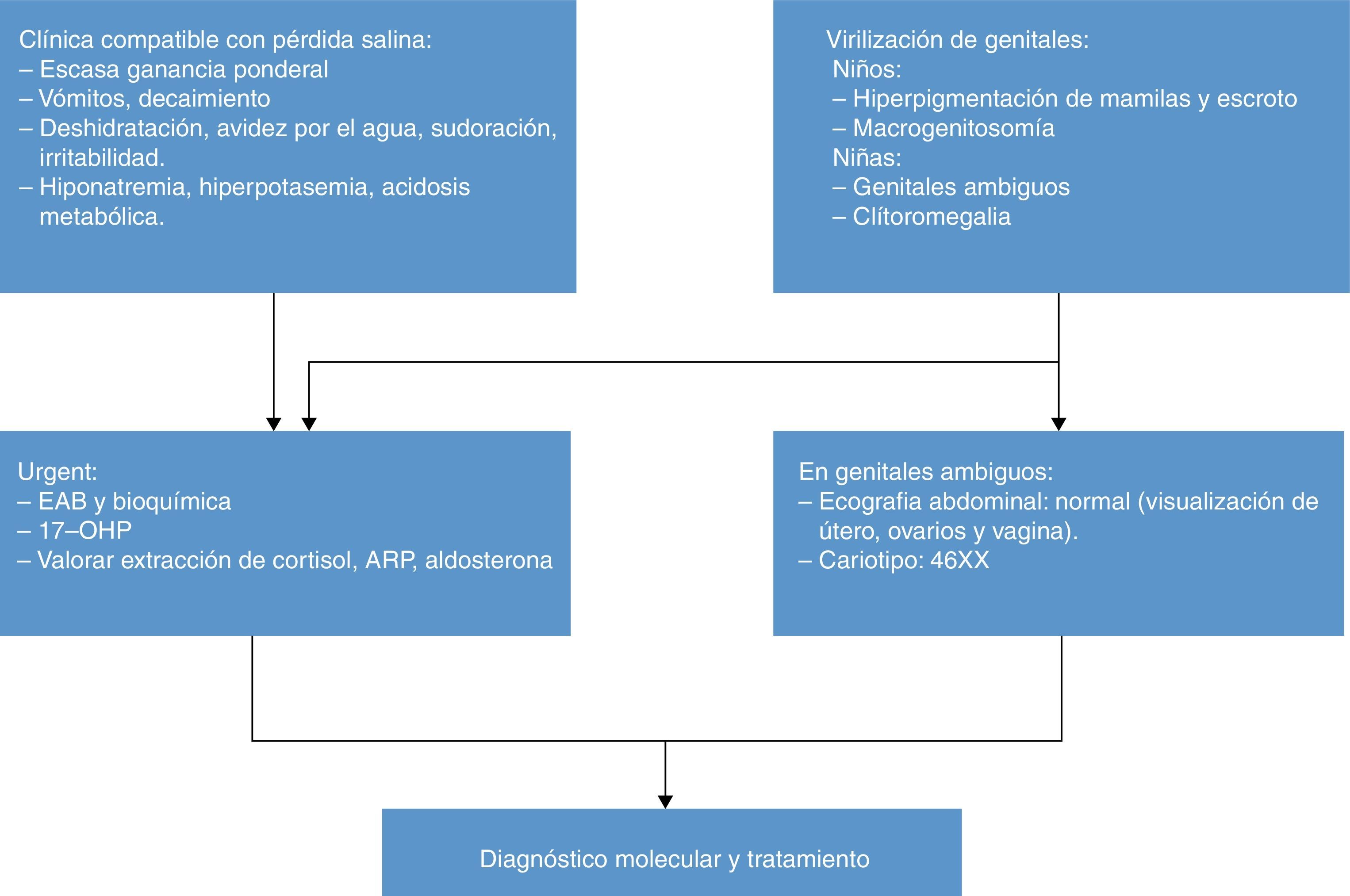
La «pérdida salina» (pérdida de peso, anorexia/avidez por el agua, vómitos, decaimiento, coma) origina hiponatremia, hiperpotasemia, tendencia a la hipoglucemia, elevación de 17OHP, androstenediona, testosterona, DHEA y ACTH, disminución de cortisol y aldosterona, y elevación de la actividad de renina plasmática (ARP).
En el periodo neonatal los inmunoanálisis directos (habitualmente utilizados en nuestro medio) muestran interferencias analíticas y pueden ocasionar elevaciones falsas de 17OHP en niños sanos o valores «normales» de aldosterona en recién nacidos con pérdida salina. El test de estimulación con ACTH no es necesario por los altos niveles de 17OHP.
El genotipado CYP21A2 es de gran utilidad para confirmar la enfermedad9,10.
El cariotipo de una paciente 21OHD con genitales ambiguos es 46XX. La ecografía abdominal y genitografía son útiles para visualizar los ovarios, las trompas y el útero.
Diagnóstico en el niño mayor/adolescenteEl diagnóstico del varón con forma virilizante simple puede verse demorado porque la clínica es inaparente en el periodo neonatal. Algunas niñas pueden presentar clitoromegalia y signos de androgenización durante la infancia. La tabla 3 recoge la pauta de actuación ante un niño/adolescente con sospecha de 21OHD.
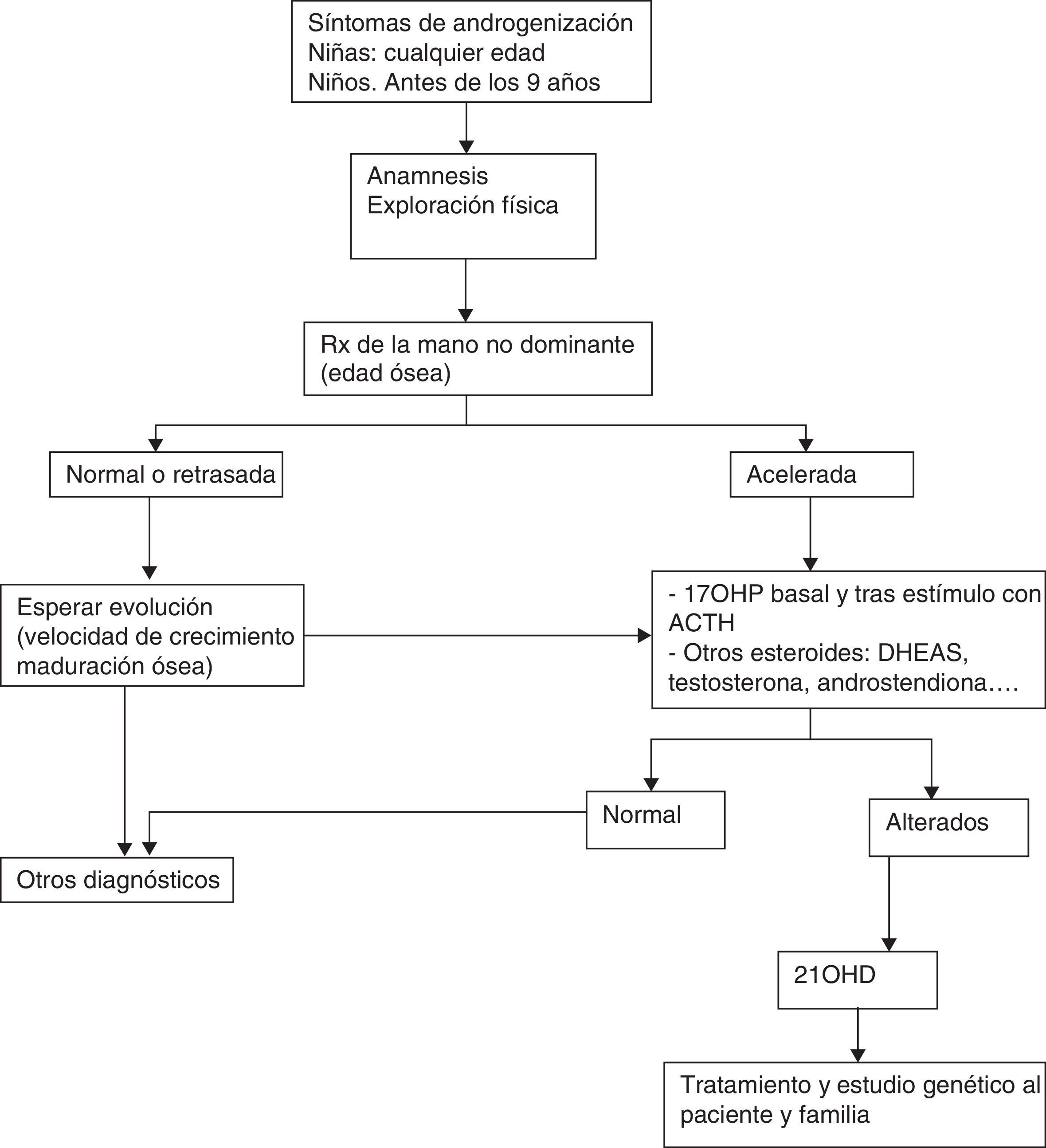
El genotipo CYP21A2 es de utilidad para discriminarlos ya que estos pacientes presentan alteraciones severas en ambos alelos.
Diagnóstico genético molecularEl gen responsable del 21OHD se denomina CYP21A2. Se localiza en el brazo corto del cromosoma 6p21.3, en la región III del sistema HLA, muy próximo al pseudogén CYP21A1P. A diferencia del gen CYP21A2, CYP21A1P presenta una serie de mutaciones que impiden su traducción en una proteína funcional, aunque ambos genes son homólogos. Todas las formas clínicas están asociadas a anomalías del gen CYP21A2; ante el diagnóstico de un paciente 21OHD es aconsejable hacer un estudio familiar que permita realizar el diagnóstico de portadores o de formas no clásicas oligosintomáticas y/o crípticas. A diferencia de otras enfermedades recesivas, en las cuales es frecuente presentar la misma mutación en los 2 alelos, en 21OHD los enfermos son frecuentemente heterocigotos compuestos, tienen mutaciones diferentes en los alelos paterno y materno, o hemicigotos, si tienen una mutación puntual en heterocigosis con una deleción en el otro alelo. Solo en el caso de mutaciones frecuentes o consanguinidad se encuentran enfermos homocigotos para una determinada mutación. Los portadores o heterocigotos presentan un solo cromosoma mutado, y en principio no manifiestan signos clínicos, aunque sí pueden presentar una respuesta elevada de 17OHP en el test de ACTH11,12.
El 95% de variantes patológicas del gen CYP21A2 son recurrentes y resultado de 2 tipos de mecanismos entre el gen CYP21A2 y el pseudogén CYP21A1, recombinación asimétrica y conversión génica. Se han descrito otras mutaciones del gen CYP21A2 no atribuibles al mecanismo de microconversión génica que representarían el 5% de los alelos. Las mutaciones se producen de novo en el 1% de casos (esto obliga a documentar la situación de portadores en los progenitores y no considerarlos portadores obligados).
La relación genotipo/fenotipo es muy intensa ya que la severidad de los signos clínicos depende del grado de déficit enzimático determinado por el tipo de afectación molecular (tabla 4) derivada de ambos alelos, y el análisis de correlación genotipo-fenotipo solo puede hacerse sobre los pacientes completamente genotipados. El fenotipo viene condicionado por el alelo más leve; las mutaciones graves pueden encontrarse en fenotipos graves y leves12.
Actividad residual de las mutaciones puntuales más frecuentes del gen de la 21-hidroxilasa, y correlación genotipo-fenotipo
| Mutación | Nomenclatura HGV NM.000500.5 | Localización CYP21A2 | Actividad residual (%)a | Forma clínicab |
|---|---|---|---|---|
| Pro30Leuc | c.92C>T (p.Pro31Leu) | Exón 1 | 30-60 | NC |
| i2 splice | c.293-13 A/C>G | Intrón 2 | <2 | PS/VS |
| Del8nt | c.332_339del | Exón 3 | 0 | PS |
| Ile72Asn | c.518T>A (p.Ile173Asn) | Exón 4 | 3 a 7 | VS |
| Cluster ex6 | c.[701T>A;713T>A;719T>A] (p.Ile237Asn;Val238Glu;Met240Lys) | Exón 6 | 0 | PS |
| Val281Leu | c.844G>T (p.Val282Leu) | Exón 7 | 20 a 50 | NC |
| InsT | c.923dupT (p.Leu308PhefsX6) | Exón 7 | 0 | PS |
| Gln318Xc | c.955C>T (p.Gln319X) | Exón 8 | 0 | PS |
| Arg356Trp | c.1069C>T (p.Arg357Trp) | Exón 8 | 2 | PS/VS |
| Arg426His | c.1280G>A (p.Arg427His) | Exón 10 | 1 a 5 | VS/PS |
| Pro453Ser | c.1360C>T(p.Pro454Ser) | Exón 10 | 20-50 | NC |
PS: pérdida salina; VS: virilizante simple; NC: no clásica; Del: deleción; Ins: inserción.
Forma clínica (PS, VS, NC) asociada cuando la alteración se presenta en homocigosis, hemicigosis o en heterocigosis compuesta con otra alteración puntual grave (actividad residual <7%).
c Debe realizarse estudio complementario de conversión génica en 5′ para Pro30 Leu en asociación con conversión 5’ es VS, o de dosis génica para Gln318X en alelo con duplicación del gen no asocia la deficiencia y no es infrecuente en la población general12.
Las mutaciones del gen CYP21A2 tienen una nomenclatura alternativa que se puede encontrar en algunas bases de datos y resulta particularmente confusa para el clínico. La razón es que la Human Genome Variation Society (HGVS) eligió para CYP21A2 una secuencia (NM00500.5) que no se correspondía con la que había sido habitualmente empleada (M12792.1) y había fundamentado el conocimiento previo, publicaciones y bases de datos de esta enfermedad. Una y otra secuencia difieren en un triplete ubicado al comienzo del exón 1 en una región polimórfica que incluye 5 o 4 CTGs, respectivamente. Es por tanto distinta la numeración de los aminoácidos a partir de dicha posición (ej. P.Pro31Leu vs. p.Pro30Leu, p.Val282Leu vs. p.Val281Leu). Posteriormente se facilitó el que se utilizara la secuencia que había sido la habitual (ENST00000448314 que se corresponde con M12792.1), aunque en la actualidad ambas denominaciones conviven.
Fuente: adaptada de Ezquieta et al.12.
En la tabla 4 se muestra la distribución de frecuencias de las mutaciones en formas clásicas y sospechas neonatales poscribado en nuestra población. En las formas graves ambos alelos presentan mutaciones severas, pero en las formas no clásicas, e incluso crípticas, podemos encontrar que ambas alteraciones sean leves o que uno de los alelos sea grave y el otro leve13.
El estudio molecular limitado a un panel que incluya las deleciones/conversiones y las mutaciones puntuales frecuentes (tabla 4) es muy informativo, 95% de alelos graves. Esta aparente «simplicidad» no debe enmascarar el hecho de que se trata de un locus complejo, cuyo análisis debe ser realizado por laboratorios con experiencia en genotipado CYP21A2.
El consejo genético es necesario en el contexto familiar de las formas clásicas y se ve resaltado por la necesidad de detectar las formas leves que presentan alteración grave, la alta frecuencia de portadores de mutación grave en población general y también la —a veces— inaparente clínica en los varones con forma virilizante.
TratamientoEl objetivo es reemplazar la secreción fisiológica de glucocorticoides y mineralocorticoides para evitar la pérdida salina, controlar los signos de hiperandrogenismo y mejorar las consecuencias en la vida adulta. En las niñas se realizará tratamiento quirúrgico de genitales externos14.
Tratamiento con glucocorticoidesLa hidrocortisona es de elección por su potencia biológica similar al cortisol endógeno y vida media corta. Prednisolona y dexametasona no se deben usar en la infancia. Se utiliza en comprimidos, triturados en niños pequeños, y no en suspensión; la distribución del fármaco en líquidos es irregular e inestable. Se debe dividir en —al menos— 3 tomas diarias, siendo difícil establecer las dosis diarias. En el recién nacido con pérdida salina las dosis son elevadas e intravenosas (50mg/m2/día) pero progresivamente van disminuyendo, siendo al año alrededor de 20mg/m2/día, en la infancia de 10-15mg/m2/día y en la adolescencia no superan los 15-20mg/m2/día3,15; siempre individualizadas al estar influenciada por múltiples factores conocidos (variabilidad clínica, genotipo, gravedad del defecto enzimático, absorción, metabolismo y farmacocinética de hidrocortisona) y desconocidos1,14.
El objetivo es tratar con la mínima dosis eficaz, que permita un paciente asintomático, un crecimiento, peso y desarrollo puberal normales y una supresión adecuada de andrógenos suprarrenales. Indican infratratamiento: aceleración en la velocidad de crecimiento, adelanto en la edad ósea, aparición de virilización. Indican sobretratamiento: deceleracion del crecimiento, retraso de edad ósea, obesidad o hipertensión arterial. Androstenediona y testosterona son los mejores indicadores de un adecuado tratamiento glucocorticoideo en pacientes prepuberales. Los cambios de tratamiento deberán realizarse en el contexto clínico de cada paciente y no solo basándose en los parámetros analíticos.
Futuras formas de administración de hidrocortisona (preparados depot, bombas de infusión) facilitarán la sustitución más fisiológica de cortisol16.
Tratamiento con mineralocorticoidesEmpleamos 9α-fluorhidrocortisona oral. Los lactantes necesitan dosis mayores en los primeros meses de vida (0,1-0,2mg/día); lactantes mayores y niños se mantienen con 0,05-0,1mg/día. En los primeros meses se administran suplementos de cloruro sódico oral (2-4mEq/kg/día) divididos en varias tomas hasta que inicien la alimentación complementaria (ClNa 20%, 1cc=3,4mEq de Na) (tabla 5).
Tratamiento de mantenimiento
| Tratamiento | Dosis total | Distribución |
|---|---|---|
| Glucocorticoides (hidrocortisona compr.) | 10-20mg/m2/día | 3 veces al día |
| Mineralocorticoides (fludrocortisona compr.) | 0,05-0,2mg/día | 1-2 veces al día |
| Suplementos de cloruro sódico (primeros meses de edad) | 2-4mEq/kg/día | En varias tomas al día |
Los pacientes con la forma virilizante simple no tienen crisis de pérdida salina, pero pueden presentar aumento de ARP (depleción crónica de sodio). Mantener el balance normal de sodio reduce los niveles de vasopresina y ACTH, contribuyendo a disminuir la dosis de hidrocortisona. Asociar en estos pacientes dosis bajas de 9α-fluorhidrocortisona facilita el tratamiento17. El tratamiento debe conseguir: ausencia de síntomas, presión arterial y frecuencia cardiaca normales, normalidad electrolítica y de ARP.
Tratamiento en las situaciones de estrésLos pacientes con formas graves no producen suficiente cortisol en situaciones de estrés, por lo que deberemos aumentar la dosis de hidrocortisona entre 2 y 10 veces en estos episodios ante el riesgo de crisis de insuficiencia suprarrenal (tabla 6). El estrés emocional, una enfermedad menor o realizar ejercicio físico no precisan mayor dosis.
Tratamiento ante situaciones de estrés
| Tratamiento de sostén | Suero salino fisiológico 10-20cc/kg si hipovolemia o shock Reponer el grado de deshidratación Corrección de las alteraciones hidroelectrolíticas (hiponatremia, hiperpotasemia, acidosis metabólica, hipoglucemia) |
| Tratamiento etiológico | Estrés leve: duplicar la dosis de hidrocortisona oral 3-4 días |
| Estrés intenso: hidrocortisona vía parenteral Dosis inicial según edad cronológica: - hasta 6 meses: 10-20mg - <2 años: 25mg - 2-12 años: 50mg - >12 años: 100mg Dosis sucesivas: 100mg/m2/día (o 3-4 veces las dosis de mantenimiento) cada 6h. Descenso a dosis habituales en cuanto la situación clínica lo permita |
Estos niños deben vacunarse según el calendario establecido.
Tratamiento quirúrgicoA diferencia de otras enfermedades que causan alteraciones en la diferenciación sexual, en estas niñas existe un amplio consenso para realizar tratamiento quirúrgico de reconstrucción hacia el sexo femenino; tienen cariotipo femenino 46XX, ovarios, trompas y útero normales y con tratamiento adecuado pueden tener un desarrollo puberal y fertilidad normal. El cirujano pediátrico diseñará la secuencia de intervenciones que deberán realizarse en centros con experiencia acreditada, de manera precoz, antes de los 24 meses para que la niña pueda establecer un esquema corporal adecuado. No se aconseja la cirugía entre los 2 años y la adolescencia en ausencia de complicaciones. Los resultados se reevaluarán en la adolescencia y edad adulta, tratando posibles complicaciones (fístulas uretrovaginales, estenosis vaginales) y valorando que la morfología de los genitales se adecúe a la anatomía femenina normal. Se deben disminuir al máximo las exploraciones genitales, siempre dirigidas a mejorar el plan de tratamiento. Si se realizan deben hacerse en presencia del menor número de personas posible y/o bajo sedación para evitar la estigmatización de la niña18,19.
Talla bajaLa hiperproducción de andrógenos adrenales y el tratamiento con dosis altas de glucocorticoides son los factores que condicionan una talla baja adulta. También pueden presentar pubertad precoz periférica y, en su evolución, pubertad precoz central siendo preciso tratar con análogos de hormona liberadora de gonadotropinas.
Los inhibidores de aromatasa (anastrazol) y la hormona de crecimiento solo se deben utilizar en pacientes con pronósticos de talla muy bajos y en estudios controlados15.
Tumores adrenalesEl amento de ACTH puede producir hiperplasia de los tejidos sensibles como glándulas suprarrenales y zonas con restos ectópicos de células adrenales (testículos y ovarios). La ecografía periódica es el método diagnóstico de elección. Requieren intensificar el tratamiento con glucocorticoides y en ocasiones cirugía20.
FertilidadEstas mujeres pueden presentar un potencial de fertilidad disminuido. El hiperandrogenismo provoca ciclos anovulatorios y alteración del eje hipotálamo-hipófiso-ovárico. Pueden presentar dispareunia por estenosis del introito vaginal y alteraciones anatómicas asociadas a la virilización o derivadas de la cirugía.
Durante el embarazo deben mantener el tratamiento previo de glucocorticoides y mineralocorticoides, y en el parto recibir hidrocortisona parenteral al ser una situación de estrés grave.
Es fundamental realizar consejo genético previo a la gestación.
Varones con formas clásicas también pueden presentar fertilidad reducida por hipogonadismo hipogonadotropo asociado y restos ectópicos adrenales testiculares. Un tratamiento deficiente facilita un aumento de andrógenos de origen suprarrenal que suprimen la FSH, disminuyendo la producción de espermatozoides2.
Alteraciones metabólicasEl tratamiento crónico con corticoesteroides es un factor de riesgo para la osteoporosis; requiere profilaxis con ejercicio físico, y aportes adecuados de calcio y vitamina D.2
La obesidad, hipertensión arterial, resistencia insulínica e intolerancia a hidratos de carbono son efectos secundarios a prevenir y tratar.
Diagnóstico y tratamiento prenatalesEl diagnóstico prenatal se plantea en el feto que presenta riesgo documentado 1/4 de padecer enfermedad grave (ambos progenitores portadores de mutaciones severas, uno de los progenitores presenta la enfermedad o ya ha habido un miembro diagnosticado en la familia). Es necesario realizar el estudio completo antes de programar un embarazo en las familias con caso índice afecto.
El objetivo del tratamiento prenatal es evitar la virilización de los genitales en el feto femenino afectado; esto supone un riesgo 1/8 en cada embarazo. Está basado en administrar dexametasona a la madre en las primeras semanas de embarazo (antes de la 7.a-8.a semanas): al atravesar la barrera placentaria es capaz de disminuir la hiperproducción de andrógenos de la glándula suprarrenal del feto, con lo que se evita la androgenización del feto femenino. Cuando se establece el diagnóstico del feto, el tratamiento se suspende en caso de que se trate de un varón o de una niña no afecta. La vellosidad coriónica es la muestra de origen exclusivo fetal más temprana, habitualmente hacia la 12.a-14.a semanas.
Este tratamiento se viene realizando desde los años 90 y es efectivo para impedir la virilización de la niña 21OHD, evitar tratamientos quirúrgicos y el impacto familiar que ocasiona el nacimiento de una niña con genitales masculinizados21,22.
Sin embargo, supone exponer innecesariamente a 7 de cada 8 fetos a dexametasona (todos los varones y 3 fetos femeninos no afectos de cada 4). La dexametasona es un fármaco teratogénico en modelos animales, con potenciales efectos secundarios a largo plazo no bien conocidos. Sobre la madre gestante puede producir algunos efectos secundarios23.
Actualmente recomendamos este tratamiento si se realiza en centros con equipo experimentado y tras consentimiento informado24.
El estudio de ADN fetal en sangre materna puede identificar el sexo a partir de la 6.a semana, aunque alcanza sensibilidad al 100% en la 10.a. No permite seleccionar los embarazos a tratar y no evita el tratamiento de niñas no afectas y portadoras. La secuenciación masiva en ADN fetal circulante posiblemente permitirá el diagnóstico muy precoz (6.a semana) del feto afecto de 21OHD, aunque todavía esta técnica se encuentra fuera de contexto clínico. La presencia conjunta de ADN fetal y materno obliga al análisis mediante marcadores polimórficos ya que la complejidad del locus CYP21A2 impide el análisis directo25.
El diagnóstico preimplantacional todavía no está disponible generalizadamente. Debe ser realizado en centros con experiencia al realizarse sobre una célula y requerir de amplificación in vitro, dadas las características especiales de este locus.
Centros clínicos de referencia (CCR)Los pacientes con formas graves deben tratarse en CCR con acreditada experiencia para evitar el tratamiento fraccionado, demorado y a veces inadecuado que pueden recibir pese a los esfuerzos que esté realizando su pediatra endocrinólogo. Deben establecerse en hospitales de máximo nivel con recursos humanos y técnicos suficientes26.
Transición a la vida adultaLos adolescentes suele faltar a las revisiones y no cumplir el tratamiento. La función gonadal adulta depende de un buen cumplimiento terapéutico; la disfunción ovárica en la mujer y los nódulos testiculares en el varón aumentan con mal cumplimiento. Temas importantes a tratar en la mujer son los relativos a su anatomía genital e historial quirúrgico. La transición a adultos debe ser consensuada y programada entre paciente, endocrinólogo pediatra y endocrinólogo27.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
M. Alonso (Hospital Ramón y Cajal, Madrid, España).
S. Berrade (Complejo Hospitalario de Navarra, Pamplona, España).
M. Bonet (Hospital del Mar, Barcelona, España).
M. Gallego (Hospital Universitario 12 de Octubre, Madrid, España).
M. Gussinyé (Hospital Vall d’Hebron, Barcelona, España).
A. Gutiérrez (Hospital del Mar, Barcelona, España).
F. Hermoso (Hospital Clínico Universitario de Valladolid, España).
J.L. Lechuga (Hospital Universitario Puerta del Mar, Cádiz, España).
C. Luzuriaga (Hospital Marqués de Valdecilla, Santander, España).
A. Oliver (Hospital Universitario La Paz, Madrid, España).
S. Quintero (Hospital Materno Insular, Las Palmas, España).
B. Roldan (Hospital Ramón y Cajal, Madrid, España).
