

Mutaciones bialélicas en el gen OTUD6B, localizado en la región 8q21.3, han sido recientemente descritas como causales de discapacidad intelectual (DI) sindrómica en 7 familias a nivel mundial1,2. Este gen codifica una enzima implicada en la desubiquitinación, proceso de retirada de la ubiquitina de las proteínas marcadas para su degradación. Las enzimas encargadas de la ubiquitinación/desubiquitinación regulan además, múltiples procesos como la señalización celular, interacciones proteína-proteína y tráfico intracellular3. Recientemente se ha relacionado la alteración de estos mecanismos con afecciones como las enfermedades autoinflamatorias4 y neurológicas5, y la desubiquitinasa OTUD6B codificada por este gen, con procesos de crecimiento celular y cáncer6.
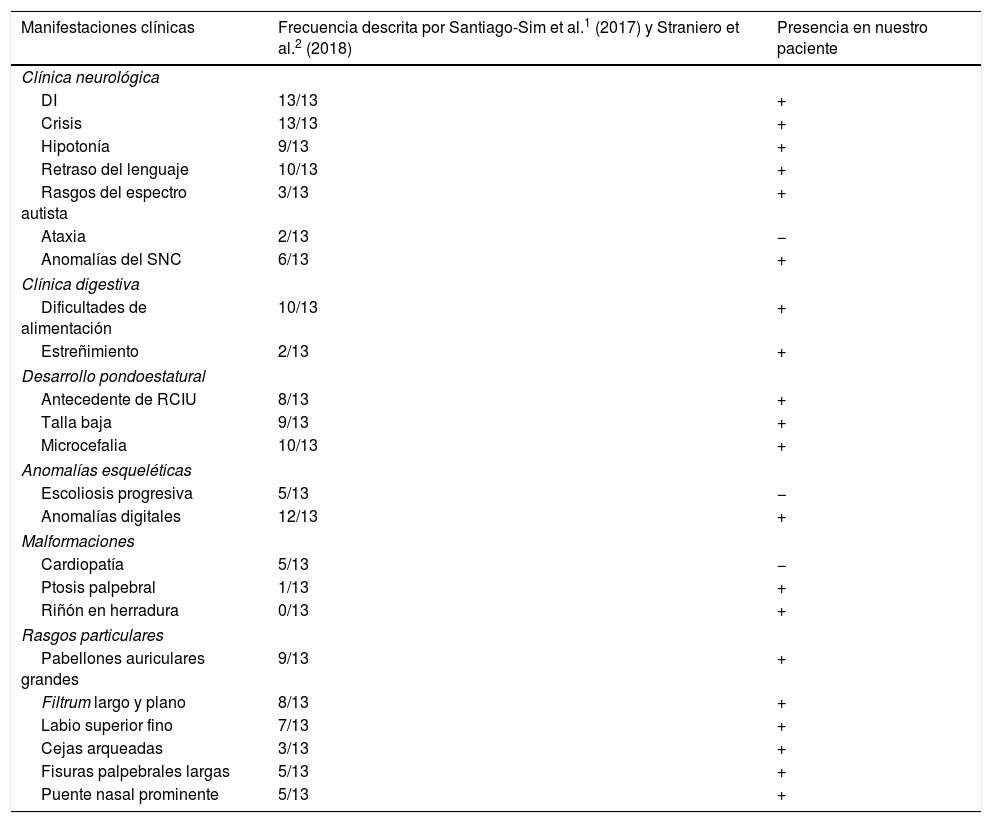
El síndrome de DI con dismorfia facial, crisis y anomalías de las extremidades (OMIM 617452), asocia en todos los casos descritos las características que lo definen, siendo habitualmente la DI grave. Otras manifestaciones frecuentemente presentes son el antecedente de un retraso de crecimiento intrauterino (RCIU), talla baja, cardiopatía y anomalías esqueléticas y neurológicas variables (rasgos del espectro autista, ataxia, etc.). Describimos por su excepcionalidad, un nuevo caso con la mutación homocigota c.433C>T en gen OTUD6B presente en 3/7 familias publicadas.
Se trata de una niña de 4 años española sin antecedentes familiares, hija de padres sanos no consanguíneos. Fue remitida a consulta de genética médica a los 3 meses por rasgos particulares, microcefalia y antecedente de RCIU. La gestación cursó sin incidencias hasta la semana 20, en la que se detectó ausencia de hueso nasal y comunicación interventricular (no confirmada). Se realizó amniocentesis resultando la qF-PCR y cariotipo normales. No presentó enfermedad perinatal y la somatometría fue normal (peso: 2.745g, p15, DE: −1,05; longitud: 48cm, p21, DE: −0,83; PC: 33cm, p19, DE: −0,9).
Evolutivamente se evidenció hipotonía generalizada y retraso global del desarrollo moderado. La sedestación se consiguió a los 11 meses, y en la actualidad presenta una deambulación inestable. Asocia rasgos del espectro autista y ausencia de lenguaje expresivo. Inició crisis tónico-clónicas febriles a los 8 meses siendo posteriormente afebriles. Los EEG y vídeo-EEG realizados han sido normales. No obstante, se ha iniciado tratamiento con ácido valproico con adecuado control de crisis. Acude a atención temprana y sigue revisiones por neuropediatría. La resonancia magnética cerebral mostró un aumento leve inespecífico del IV ventrículo sin otras anomalías asociadas.
Durante los primeros meses comenzó con reflujo gastroesofágico moderado, precisando tratamiento con omeprazol, y problemas de deglución. En la actualidad toma todos los alimentos triturados. No ha precisado cirugías ni alimentación por sonda nasogástrica. Asocia estreñimiento controlado con dieta.
En cuanto al desarrollo pondoestatural presenta talla baja (95cm, p2, DE: −2,1) y microcefalia moderada (PC: 45cm, p<1, DE: −4,21). Durante su seguimiento se realizó despistaje de otras posibles anomalías congénitas asociadas, detectándose riñones en herradura, por lo que sigue revisiones en nefrología. La función renal es normal.
También ha sido valorada por oftalmología por ptosis izquierda con estrabismo leve asociado, detectándose miopía, por lo que porta lentes. La audición es normal. A nivel esquelético destaca un cuello corto, actitud cifótica, pies planos con talón prominente, limitación de la extensión de codos y rodillas y anomalías digitales consistentes en ensanchamiento de falanges distales, clinodactilia de quinto dedo bilateral y primer dedo ancho en manos y pies, y superposición de quinto dedo sobre cuarto con acortamiento de segundo dedo en ambos pies. En la tabla 1 se compara la clínica de nuestra paciente con la descrita en la literatura.
Características clínicas de los casos publicados
| Manifestaciones clínicas | Frecuencia descrita por Santiago-Sim et al.1 (2017) y Straniero et al.2 (2018) | Presencia en nuestro paciente |
|---|---|---|
| Clínica neurológica | ||
| DI | 13/13 | + |
| Crisis | 13/13 | + |
| Hipotonía | 9/13 | + |
| Retraso del lenguaje | 10/13 | + |
| Rasgos del espectro autista | 3/13 | + |
| Ataxia | 2/13 | − |
| Anomalías del SNC | 6/13 | + |
| Clínica digestiva | ||
| Dificultades de alimentación | 10/13 | + |
| Estreñimiento | 2/13 | + |
| Desarrollo pondoestatural | ||
| Antecedente de RCIU | 8/13 | + |
| Talla baja | 9/13 | + |
| Microcefalia | 10/13 | + |
| Anomalías esqueléticas | ||
| Escoliosis progresiva | 5/13 | − |
| Anomalías digitales | 12/13 | + |
| Malformaciones | ||
| Cardiopatía | 5/13 | − |
| Ptosis palpebral | 1/13 | + |
| Riñón en herradura | 0/13 | + |
| Rasgos particulares | ||
| Pabellones auriculares grandes | 9/13 | + |
| Filtrum largo y plano | 8/13 | + |
| Labio superior fino | 7/13 | + |
| Cejas arqueadas | 3/13 | + |
| Fisuras palpebrales largas | 5/13 | + |
| Puente nasal prominente | 5/13 | + |
DI: discapacidad intelectual; RCIU: retraso de crecimiento intrauterino; SNC: sistema nervioso central.
Presenta rasgos particulares que han cambiado evolutivamente, como apreciamos en la figura 1. Los padres firmaron un consentimiento informado para la publicación de las fotografías.

Tras su primera valoración se solicitó arrayCGH de 60k que fue normal. Ante la sospecha de los síndromes Nicolaides-Baraitser y de Baraitser-Winter cerebro-fronto-nasal, se realizaron estudios de secuenciación de genes asociados mediante técnicas de secuenciación de nueva generación, sin encontrar ninguna mutación. Finalmente se realizó exoma, que mostró la variante homocigota c.433C>T previamente descrita en OTUD6B1, confirmando a los progenitores como portadores.
Como conclusiones, describimos el primer caso en España, cuya clínica solapa con la previamente descrita y amplía el fenotipo al asociar riñón en herradura y miopía.
Este nuevo caso corrobora la implicación del gen OTUD6B en la DI sindrómica y muestra una vez más la gran utilidad del exoma para diagnosticar fenotipos complejos, recientemente descritos y de extremada baja frecuencia, que solapan con distintos síndromes clásicos y que están probablemente infradiagnosticados por su desconocimiento.
La variante coincide con la descrita en 3/7 familias publicadas, lo que sugiere que se trata de una mutación recurrente.
Debe incluirse en el diagnóstico diferencial de los defectos de glicosilación, el síndrome de Nicolaides-Baraitser y el síndrome de Baraitser-Winter, y de los ya sugeridos, síndrome de Rubinstein-Taybi y síndrome de Kabuki.
