


Se presenta un paciente varón de cuatro años de edad procedente de Gambia, con antecedentes de múltiples fracturas. Existe una consanguinidad de segundo grado entre sus padres, siendo tío y sobrina, presentando una hermana mayor de 15 años con el mismo cuadro clínico e historial médico de numerosas fracturas. El paciente se encuentra en seguimiento en consultas de Endocrinología y Traumatología Pediátrica, con el diagnóstico de osteoporosis pseudogliomatosa con mutación confirmada en homocigosis c.4015_4106delAT en el gen LRP5 (OMIM*603506).
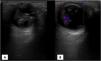
Durante el periodo neonatal se le diagnosticó vítreo primario hiperplásico bilateral, que derivó en un deterioro progresivo e irreversible de la función visual (fig. 1A y 1B).
A los dos años de vida presentó una fractura diafisaria de húmero proximal derecho, desplazada tras un pequeño golpe que precisó de reducción ortopédica (fig. 2A). Al año siguiente, otro traumatismo menor le originó una fractura de la meseta tibial izquierda sin desplazamiento, que fue tratada mediante inmovilización y tratamiento conservador (fig. 2B). A los cuatro años de edad sufrió una caída que le provocó una fractura subtrocantérea del fémur izquierdo (fig. 2C).
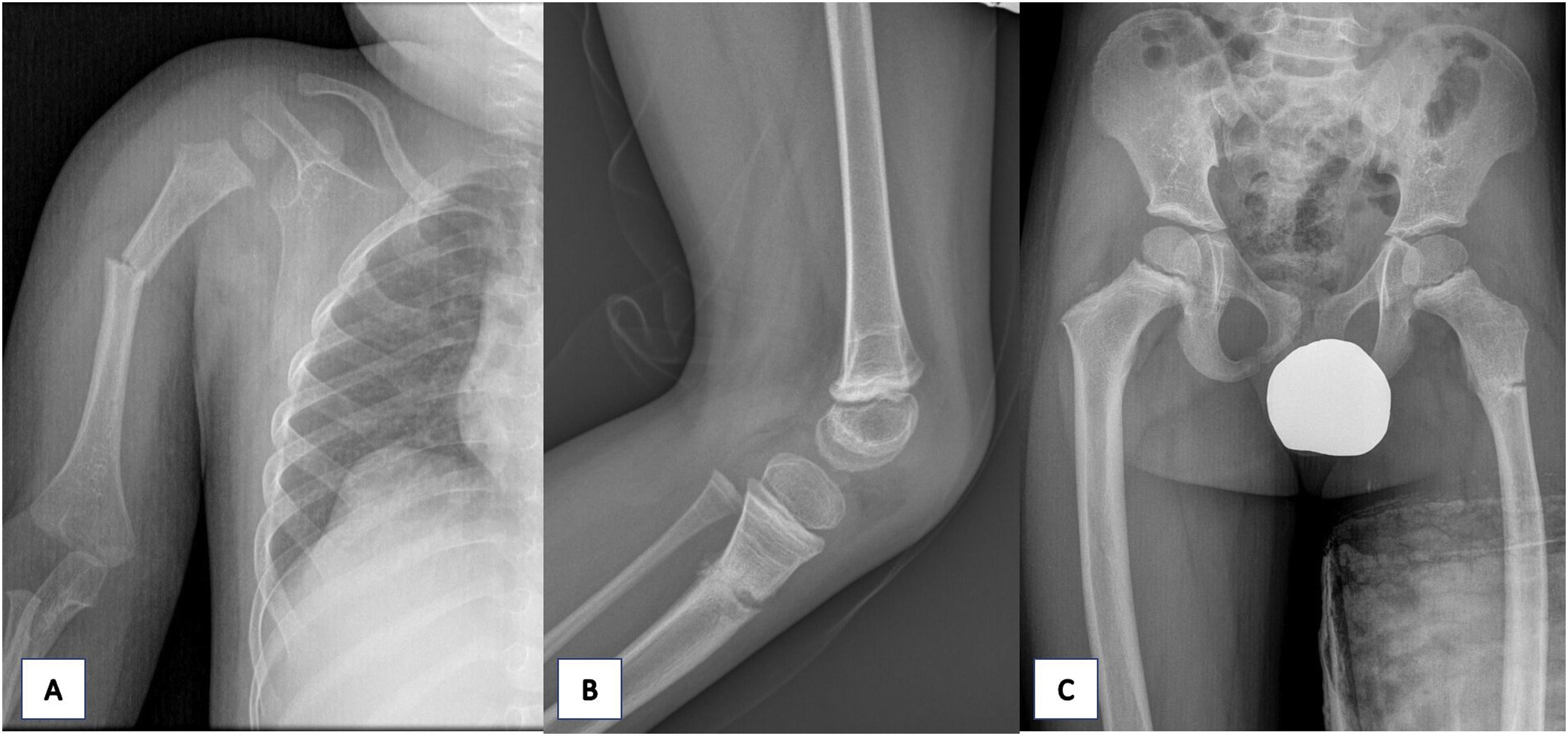
Radiografía AP húmero derecho. A) Se visualiza disrupción y angulación cortical de la diáfisis proximal correspondiente con fractura desplazada. Radiografía lateral rodilla izquierda. B) Se objetiva fractura metafisaria de la tibia. Radiografía de pelvis AP. C) Se pone de manifiesto la disrupción y radiolucencia cortical correspondiente con fractura subtrocantérea del fémur izquierdo sin desplazamiento.
El síndrome de osteoporosis pseudogliomatosa es un trastorno genético con patrón de herencia autosómico recesivo poco común, con una prevalencia de 1/2.000.0001, caracterizado por una osteoporosis grave y ceguera de aparición temprana. Las mutaciones de pérdida de función en el gen que codifica la proteína 5 relacionada con el receptor de lipoproteínas de baja densidad (LRP5), se han establecido como el defecto genético de la enfermedad2. Como diagnósticos diferenciales hay que tener en cuenta fundamentalmente las formas severas y moderadas deformantes de la osteogénesis imperfecta, y otras menos conocidas como el síndrome de Cole-Carpenter o el síndrome de Antley-Bixler, que cursa con múltiples malformaciones faciales, esqueléticas y urogenitales3.
El proceso osteopénico parece estabilizarse con la edad, por lo tanto, la profilaxis y el cuidado de las fracturas y deformidades son necesarios para prevenir problemas incapacitantes a largo plazo.
FinanciaciónEste trabajo no ha recibido ningún tipo de financiación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
