




La obstrucción de las fosas nasales en el neonato es un cuadro potencialmente fatal por su respiración nasal obligada. La causa principal es inflamatoria o infecciosa, y con menor frecuencia puede ser de origen congénito, neoplásico, traumático o iatrogénico.
La atresia de coanas es la anomalía congénita nasal más común. Una etiología menos frecuente de obstrucción nasal congénita es la estenosis de la apertura piriforme. Debe pensarse en esta última en todo recién nacido con cornaje y dificultad respiratoria de grado variable, asociado a la dificultad de pasar una sonda a través de la región anterior de las fosas nasales. El diagnóstico se confirma por tomografía computarizada del macizo craneofacial. La conducta terapéutica dependerá de la gravedad de los síntomas.
Describimos nuestra experiencia con 5 pacientes que presentaban esta afección, tratados quirúrgicamente mediante abordaje sublabial y colocación de tutor nasal.
Nasal obstruction in neonates is a potentially fatal condition due to their exclusive nasal breathing. The main cause is inflammatory or infectious rhinitis. Congenital, neoplastic, traumatic or iatrogenic causes are less frequent.
Choanal atresia is the most common congenital nasal anomaly. A less common etiology of congenital nasal obstruction is pyriform aperture stenosis. Suspicion might arise in any newborn with varying degrees of stridor and respiratory distress, associated with the difficulty of passing a probe through anterior nares. Diagnosis should be confirmed by a computed tomography of the craniofacial massif. The therapeutic approach will depend on the severity of symptoms.
We describe our experience with 5 patients with this condition, treated surgically using a sub-labial approach, and followed by nasal stenting.
La apertura piriforme (AP) es la porción ósea más anterior y estrecha de las fosas nasales. Su estenosis congénita es una causa poco frecuente de obstrucción en el neonato, producto del crecimiento óseo excesivo del proceso nasal medial del hueso maxilar de forma bilateral1,2. La incidencia es desconocida3. Puede presentarse aislada o asociada a malformaciones craneofaciales o nerviosas centrales.
El síntoma más habitual es el cornaje, que es el ruido generado por la alteración del flujo de aire al pasar por las fosas nasales de calibre disminuido3. Otros posibles síntomas son la dificultad respiratoria, los trastornos de la deglución, las apneas y la cianosis cíclica que mejora con el llanto3,4. Al examen físico existe imposibilidad o dificultad de pasar una sonda más allá del vestíbulo nasal3. El diagnóstico se confirma por tomografía computarizada (TC) del macizo craneofacial, donde se observa la disminución del área nasal a nivel de la entrada ósea5,6.
En caso de dificultad respiratoria moderada a grave, trastorno de la deglución y ante el fracaso de métodos conservadores, el tratamiento es quirúrgico y consiste en el ensanchamiento de la AP mediante abordaje sublabial, procedimiento que es seguro y eficaz3,5,7.
Casos clínicosPresentamos 5 pacientes con estenosis congénita de la apertura piriforme (ECAP) tratados por el Servicio de Otorrinolaringología Infantil entre octubre de 2009 y mayo de 2013 (tabla 1).
Pacientes con estenosis congénita de la apertura piriforme
| Paciente | Sexo | Diámetro transverso máximo de la AP | Edad al diagnóstico | Edad en el momento de la cirugía | Malformaciones asociadas |
| 1 | M | 3,50mm | 1 mes | 2 meses | Incisivo superior único central |
| 2 | M | 5,97mm | 3 meses | 4 meses | - |
| 3 | M | 4,47mm | 3 meses | 4 meses | Síndrome de Pfeiffer |
| 4 | F | 5,34mm | 2 meses | 2 meses | Esquizencefalia, incisivo superior único central |
| 5 | M | 4,83mm | 1 mes | 1 mes | Diabetes insípida, incisivo superior único central |
AP: apertura piriforme; F: femenino; M: masculino.
Los pacientes habían nacido a término y presentaron obstrucción nasal desde el nacimiento.
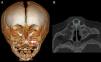
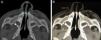
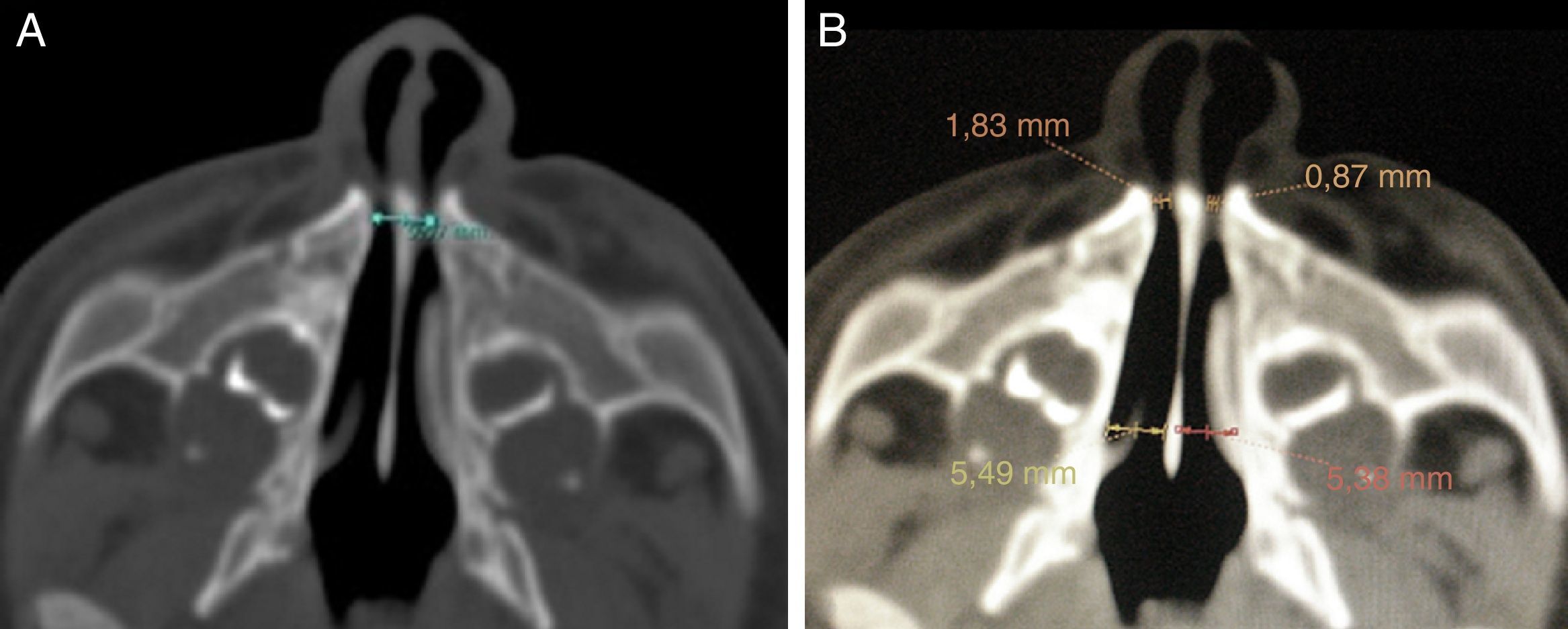
La exploración física reveló un vestíbulo nasal estrecho e imposibilidad de pasar una sonda K30 (diámetro 2,8mm) hacia la fosa nasal. La TC evidenció estenosis ósea de la AP a expensas de ambas ramas ascendentes del hueso maxilar, con calibre de coanas normal, y en 3 pacientes, un megaincisivo superior central (fig. 1). En todos los casos se midió el diámetro transverso máximo de la AP entre la cara medial del hueso maxilar a nivel del meato inferior, en el corte axial, siendo la media de 4,82mm (fig. 2). A 2 pacientes se les realizó una tomografía helicoidal con reconstrucción tridimensional (3D), que mostró la disminución de la luz nasal.
Se efectuó resonancia magnética cerebral y evaluación genética y endocrinológica. Un paciente presentaba esquizencefalia, otro, diabetes insípida central, y un tercero, síndrome de Pfeiffer (enfermedad genética rara que se caracteriza por craneosinostosis, hipoplasia media facial, sindactilia y pulgares gruesos).
Los 5 pacientes fueron tratados inicialmente con descongestivos y esteroides nasales (fenilefrina y dexametasona) por 10 días, con humidificación y aspiración de secreciones según necesidad, y se los alimentó por sonda orogástrica. Por la gravedad de los síntomas y el fracaso del tratamiento médico, se decidió la corrección quirúrgica.
Se obtuvo una vía aérea estable en todos ellos previamente al tratamiento quirúrgico mediante chupete de McGovern.
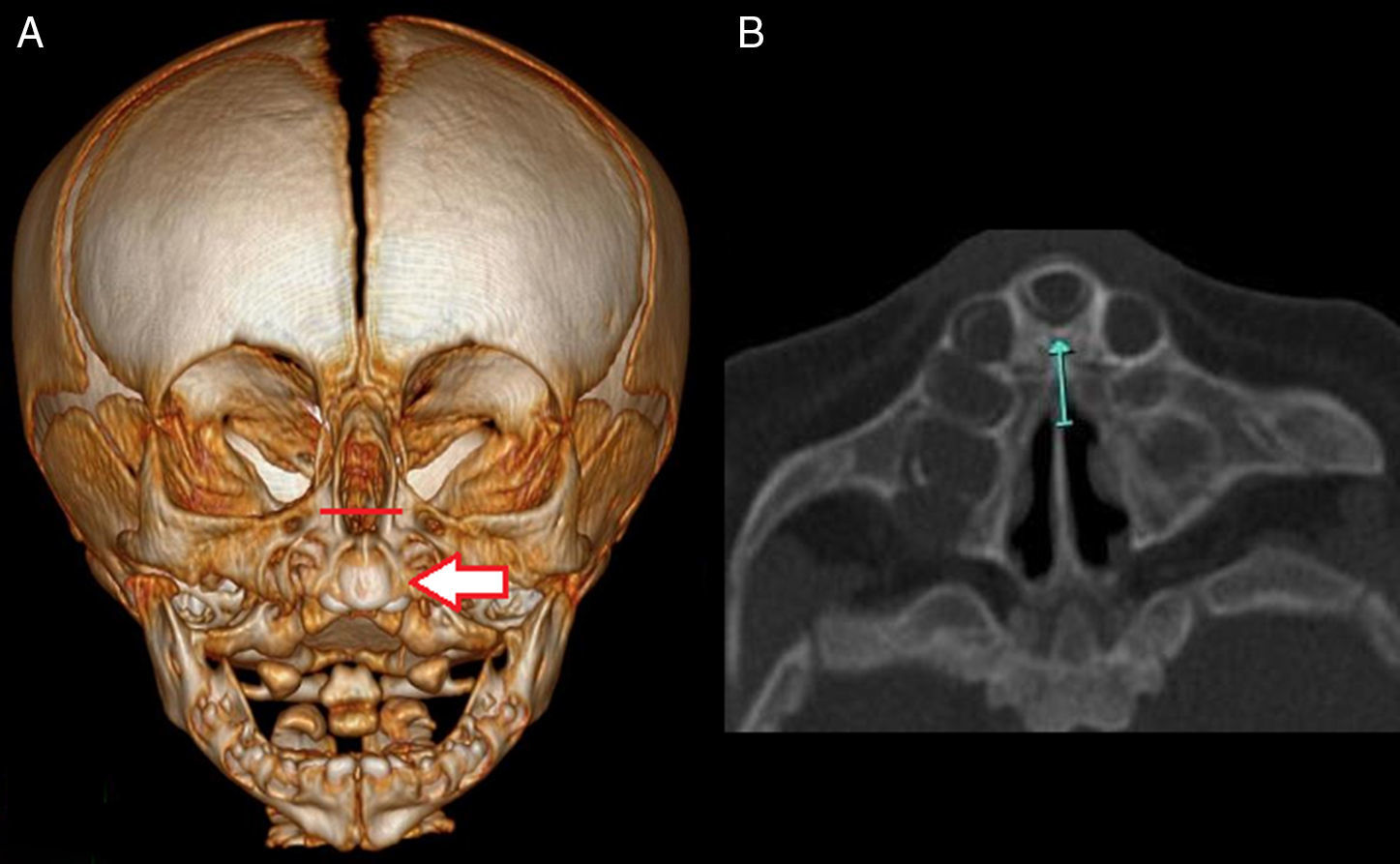
El procedimiento quirúrgico consistió en la ampliación de la AP por acceso sublabial con preservación de la mucosa nasal mientras se fresaba la rama ascendente del maxilar y el piso de la fosa. En todos los pacientes se colocó un tutor siliconado bilateral (tubo endotraqueal n.o 3,5: diámetro externo 5,3mm) como sostén de la luz endonasal durante 7 días (fig. 3).
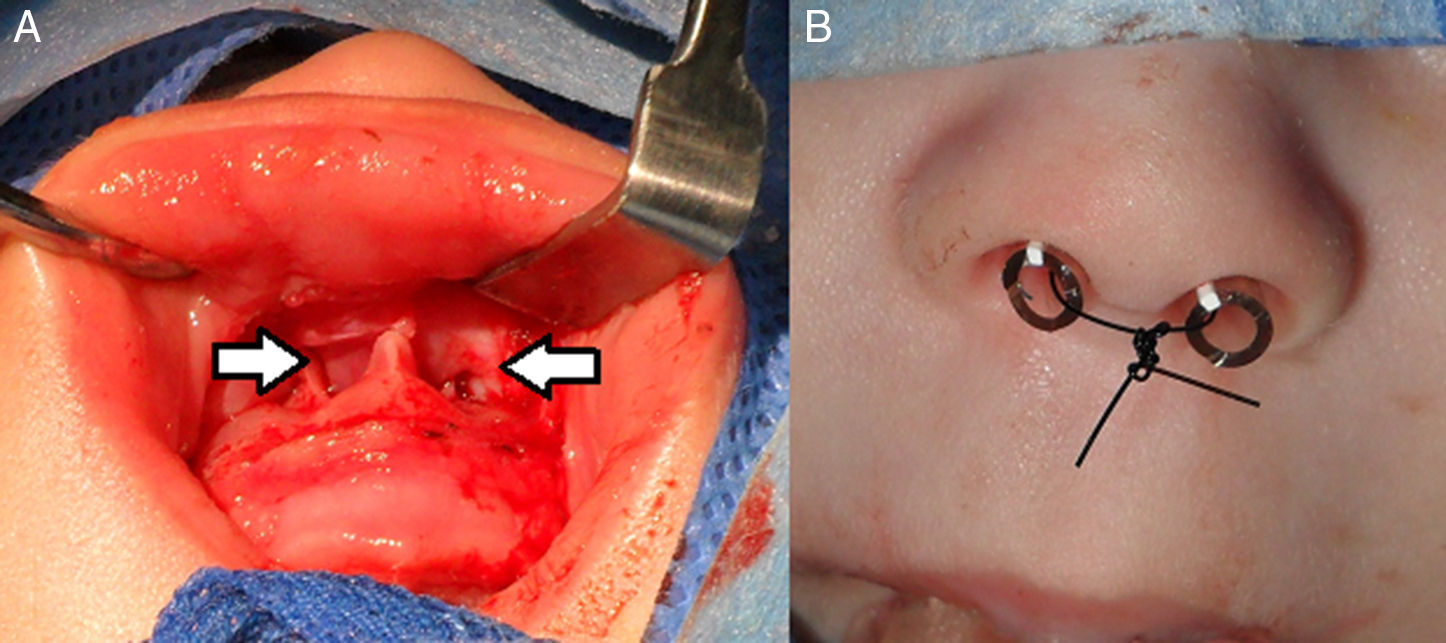
Se indicó alimentación por sonda orogástrica hasta probar tolerancia oral y, durante el tiempo de permanencia del tutor nasal, lavado nasal con solución fisiológica a través del tutor, aspiración de secreciones según necesidad y mupirocina nasal para evitar lesiones por decúbito del mismo. El alta hospitalaria fue otorgada entre los 7 y 10 días posquirúrgicos.
A los 3 meses de la cirugía de ampliación, el paciente con síndrome de Pfeiffer fue traqueotomizado por apneas obstructivas.
El tiempo medio de seguimiento fue de 22 meses. Se logró ventilación nasal adecuada en 4 pacientes, con crecimiento facial normal y tamaño del vestíbulo nasal apropiado para la edad. Un paciente, con fosas nasales permeables, permanece con traqueotomía por glosoptosis.
DiscusiónLa ECAP es una causa infrecuente de obstrucción nasal en el neonato. El diagnóstico precoz y el tratamiento apropiado son esenciales debido a su respiración nasal obligada.
La presentación clínica de la estenosis bilateral de coanas y de la ECAP es similar, pero la estenosis y la atresia coanal son más frecuentes y conocidas3,6.
En el examen físico, la rinoscopia anterior es dificultosa debido a la escasa luz que se observa por la proyección medial de la mucosa que recubre al hueso de la pared lateral de la fosa nasal. La introducción de una sonda de aspiración K30 (2,8mm de diámetro) no progresa más allá de 1cm3.
El diagnóstico de certeza se realiza mediante una TC del macizo craneofacial, donde se observa la disminución del diámetro del área nasal a nivel de la AP, mientras que las coanas son de calibre normal. El diámetro transverso de cada orificio piriforme menor de 3mm o un diámetro transverso total menor de 11mm, en recién nacidos a término, confirma el diagnóstico1,5,8. Otros hallazgos radiológicos que se asocian frecuentemente son alteraciones en la dentición (megaincisivo central único) y paladar duro triangular con una cresta en la línea media5,6.
La tomografía 3D permite una visión completa de la AP en su plano frontal, lo que posibilita medir su amplitud y una definición exacta del área ósea que debe ser resecada2,3.
Una vez descartada la rinitis inflamatoria o infecciosa, que es la causa más frecuente de obstrucción nasal en neonatos y lactantes, debe considerarse la ECAP como diagnóstico diferencial, junto a la estenosis o atresia de coanas, desvío septal, encefalocele y tumores nasales, como gliomas o quistes dermoides1–3.
La presencia de anomalías asociadas debe ser estudiada mediante endoscopia nasal, resonancia magnética de cerebro y evaluación genética y endocrinológica. Se ha descrito la asociación con megaincisivo o incisivo único central maxilar, holoprosencefalia, agenesia hipofisaria, disgenesia tiroidea y alteraciones cromosómicas, entre otros1,2,4,7–9.
Es fundamental evaluar otros sitios de obstrucción de la vía aérea, el tono muscular y la necesidad de traqueotomía. En uno de nuestros pacientes la corrección quirúrgica nasal fue exitosa, pero no se pudo lograr la decanulación por presentar glosoptosis.
La conducta terapéutica dependerá de la insuficiencia respiratoria y de su repercusión sobre el desarrollo ponderal del infante5,10.
En primer lugar, se debe establecer una vía aérea segura mediante chupete de McGovern, cánula oral o intubación orotraqueal7. La traqueotomía se reserva a las situaciones en que otras anomalías craneofaciales están asociadas.
Los casos leves se tratan de forma conservadora con descongestivos locales y humidificación hasta que la cavidad nasal crezca y la obstrucción desaparezca3.
Cuando el tratamiento quirúrgico está indicado, la técnica más aceptada es la ampliación de la AP mediante un acceso sublabial. La disección y el fresado deben realizarse de forma anterior al cornete inferior, para evitar el daño del conducto nasolagrimal. Se debe prevenir el daño de la mucosa nasal y de los brotes dentales en el piso3,7. Posteriormente pueden colocarse o no tutores nasales. Se ha recomendado el uso de tutor intraluminal de Silastic® durante el posoperatorio, para prevenir la reestenosis y favorecer la irrigación de solución salina y la aspiración de secreciones3,7.
Si bien la ECAP es poco frecuente, se la debe incluir en el diagnóstico diferencial de obstrucción de las vías respiratorias en neonatos y lactantes. Los pacientes deben ser estudiados para identificar posibles malformaciones asociadas. El tratamiento clínico quirúrgico se basa en la gravedad de los síntomas.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
