



La morfea o esclerodermia cutánea localizada es una entidad de causa desconocida1–3, que aparece raramente en la edad pediátrica3,4. Esta forma de esclerodermia es más frecuente que la cutánea generalizada y que la sistémica en el niño5. El pico de mayor incidencia tiene lugar entre los 20–40 años, aunque hasta un 15% comienza antes de los 10 años1,2. Las formas congénitas ya están presentes al nacimiento, pero su diagnóstico suele retrasarse varios años, como reflejan los casos publicados hasta el momento, que han sido diagnosticados en niños mayores o en adultos jóvenes4.
Se presenta el caso de una niña en la que, desde la primera exploración tras el nacimiento, se observaba una lesión macular en la parte izquierda del abdomen. En su primera revisión rutinaria en el centro de salud a los 15 días de vida la lesión adquiría una coloración parduzca y su tamaño era de unos 6×2cm. No presentaba antecedentes de enfermedades cutáneas similares en la familia, ni de enfermedades reumáticas o inmunológicas. El embarazo y el parto habían sido de curso normal, sin antecedentes de ingesta de fármacos, enfermedades durante la gestación o traumatismos durante el parto. No presentó en ningún momento otros síntomas asociados.
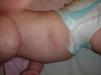
Se mantuvo en observación y en posteriores revisiones la lesión persistía, creciendo a la par o algo superior a la velocidad de crecimiento de la superficie corporal. A los 4 meses se objetivaba una lesión de 10×6cm, de contorno oval, aspecto algo pigmentado y telangiectásico, ligeramente deprimida respecto a la piel normal circundante e impellizcable al tacto, sospechándose una morfea congénita (fig. 1), por lo que se deriva a la consulta de dermatología.

Se inició tratamiento con metilprednisolona aceponato en oclusión plástica una vez al día durante un mes, asociada a calcipotriol 2 veces al día, hasta completar 6 meses. A los 18 meses de edad (fig. 2), la lesión era de unos 6,5×3cm y la pigmentación se iba atenuando progresivamente, persistiendo cierta atrofia y esclerosis cutánea. No se han desarrollado complicaciones sistémicas.

La morfea congénita se presenta ya al nacimiento, por lo que es frecuente que las lesiones que se manifiestan a la inspección sean confundidas con otras propias del período neonatal. Su diagnóstico es fundamentalmente clínico5, suele presentarse como zonas eritematosas que se tornan amarillas o blancas con un halo violáceo; posteriormente evolucionan a zonas de atrofia y en ocasiones, se atenúan, se fibrosan y pueden dejar marcas residuales de hipo o hiperpigmentación3. En la mayor parte de los casos la realización de un estudio histológico no es necesario, en especial en aquellos pacientes en los que el diagnóstico tiene lugar en la edad pediátrica3,5. Si en caso de duda clínica, se realizase una biopsia cutánea, los hallazgos histológicos consistirían en un importante engrosamiento de los haces de colágeno de la dermis. Las pruebas complementarias no suelen estar alteradas, salvo en algunos casos donde se pueden encontrar reactantes de fase aguda o autoanticuerpos elevados3, sin que esto implique un cambio en el pronóstico, por lo que tampoco sería imprescindible su realización para el diagnóstico. Debemos tener en cuenta en el diagnóstico diferencial otras entidades como el «stiff skin síndrome», nevus congénito gigante, manchas café con leche o la mastocitosis1,4.
El tratamiento va a variar en función del subtipo, de la localización y de la extensión de las lesiones. En las formas localizadas y leves se puede optar por un tratamiento conservador sin fármacos o empleando corticoides tópicos (con o sin oclusión)3. En algunos casos es preciso emplear otros tratamientos, entre los cuales se encuentran: metotrexato, calcipotriol, corticoides sistémicos, calcitriol, UVA, terapia física, cirugía y apoyo psicológico3,6. La actividad de la lesión cutánea suele durar entre 3–5 años1,5. En el seguimiento de estos pacientes podemos observar con frecuencia lesiones residuales en forma de hipo o hiperpigmentación1–3.
