


La displasia epitelial intestinal (DEI) es una causa rara de diarrea intratable1. Habitualmente, se presenta en el periodo neonatal como una diarrea grave de carácter secretor que produce fracaso intestinal permanente con dependencia indefinida de nutrición parenteral (NP). Aunque se describen casos con buena evolución2, la mayoría son dependientes de NP, por lo que el trasplante intestinal es una alternativa terapéutica útil para conseguir la autonomía digestiva3.
Se revisa de forma retrospectiva a 3 pacientes diagnosticados de displasia epitelial. Se analizan variables epidemiológicas, clínicas, diagnósticas, terapéuticas y evolutivas.
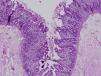
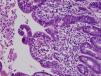
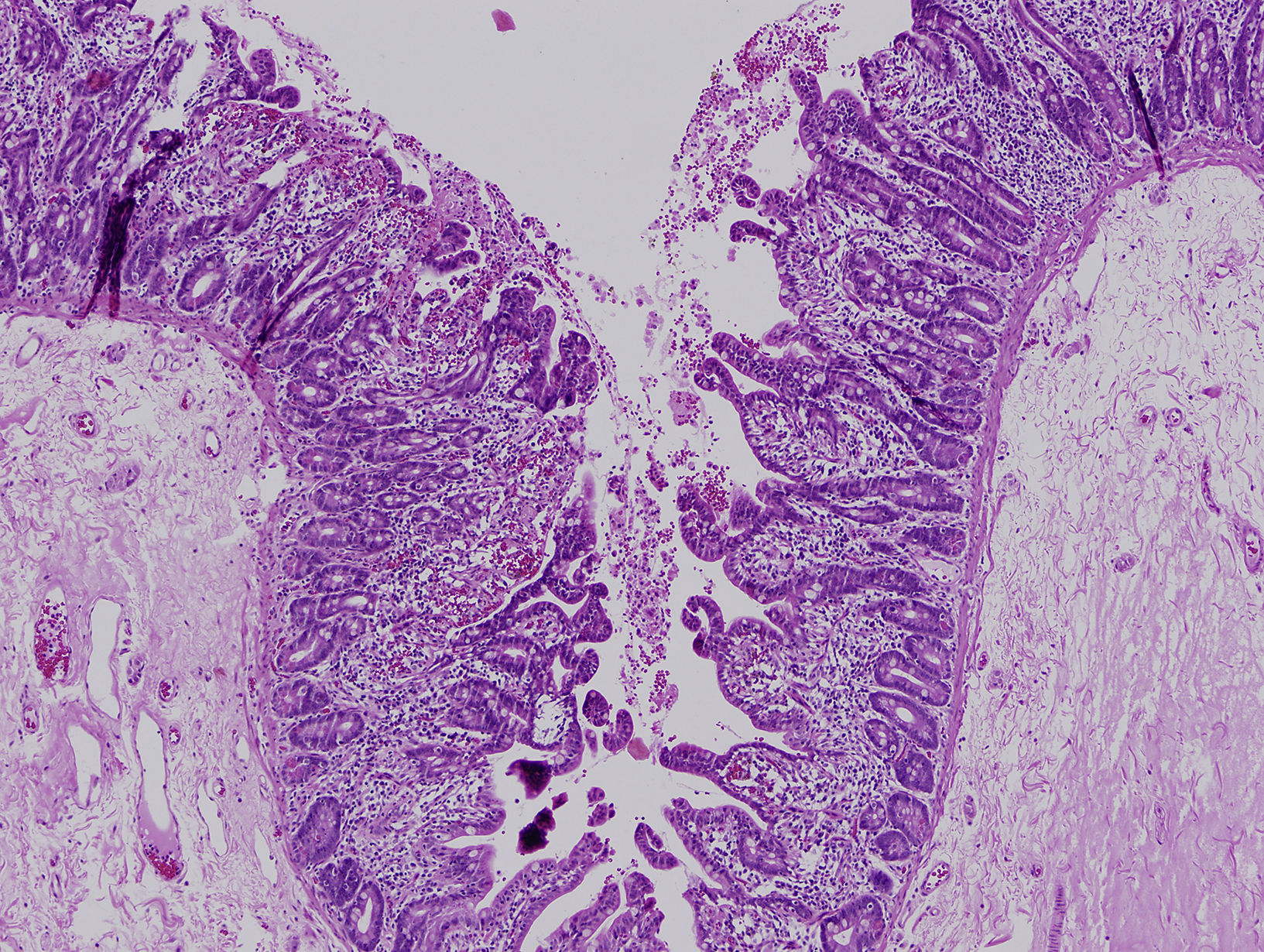
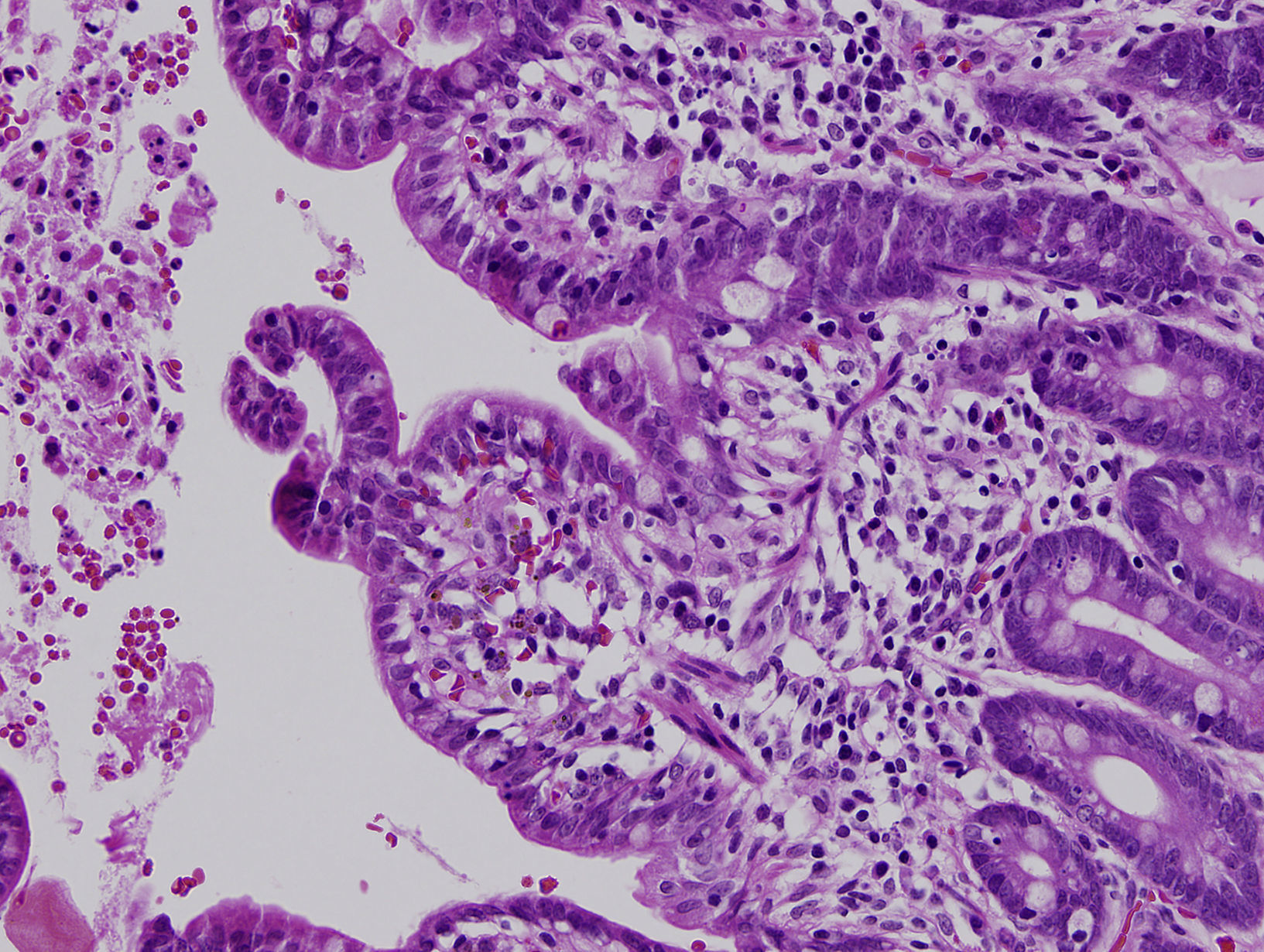
La diarrea secretora comenzó en el primer mes de vida en los 3 pacientes. Una era mujer y 2, varones. La primera paciente era de nacionalidad ecuatoriana y los 2 últimos eran hermanos, de origen marroquí y nacidos de padres consanguíneos. Todos fueron recién nacidos a término y presentaron un peso adecuado al nacimiento. Las ecografías prenatales fueron normales. Ninguno de los pacientes presentó atresia ni queratitis puntiforme. El estudio inmunológico fue normal. El estudio histológico de la mucosa intestinal demostró acortamiento de las vellosidades y agrupaciones de enterocitos de que formaban penachos (figs. 1 y 2). En el segundo y tercer paciente se confirmó la existencia de mutación en el gen EpCAM en homocigosis. Se detectó una deleción intragénica de 17 bases (c.352_368del, NM_002354,2). No se realizó estudio genético en la primera paciente.
Todos precisaron NP para evitar la deshidratación. La indicación del trasplante intestinal se estableció en la primera paciente ante el estado de malnutrición severa y altos requerimientos de electrolitos. Y en el segundo y tercer paciente cuando aparecieron las complicaciones de la NP, sepsis y hepatopatía.
Actualmente, los 3 pacientes sobreviven. La primera paciente es portadora de un trasplante multivisceral tras 8 años, el segundo paciente es portador de un trasplante multivisceral tras 7 años y el tercer paciente se encuentra pendiente de revaloración terapéutica tras pérdida del injerto por un rechazo exfoliativo. Los 2 primeros pacientes tienen alimentación oral exclusiva, curva ponderal ascendente y buena calidad de vida.
La DEI fue descrita por primera vez en 1994 por Reifen et al.1. La prevalencia se estima 1/50.000-100.000 nacidos, siendo mayor en áreas de origen árabe4. En algunos casos existe antecedente de hermanos fallecidos en los primeros meses de vida por diarrea grave. Han sido reportados varios casos desde la descripción de esta entidad, pero ninguno en nuestro país. Se caracteriza por una diarrea grave de características secretoras que comienza en los primeros días de vida4. Se produce un cuadro de malabsorción con fallo de medro.
El diagnóstico se basa en la demostración de los hallazgos histológicos típicos de esta enfermedad. Al microscopio óptico se objetiva una atrofia vellositaria intensa, con hiperplasia críptica y ausencia de infiltrado inflamatorio en la lámina propia. El hallazgo característico son los acúmulos de enterocitos empaquetados, que confieren un borde romo a la vellosidad y constituyen los penachos a que alude el nombre de esta entidad1. En la microscopia electrónica los hallazgos ultraestructurales que acompañan a los penachos de enterocitos son un discreto acortamiento del borde en cepillo y un aumento en el número y la longitud de los desmosomas entre enterocitos. Mediante la inmunohistoquímica MOC31 se tiñe, en condiciones normales, el epitelio de la mucosa, sin observarse tinción de la lámina propia ni de los vasos. En los casos de DEI existe una ausencia completa de tinción de la superficie epitelial, criptas y penachos. Algunos pacientes asocian rasgos dismórficos, atresia esofágica, atresia de coanas y anorrectal4. Y hasta el 60% de los pacientes presenta queratitis puntiforme4.
Sivagnanam et al. identificaron una mutación en el gen EpCAM, localizado en el cromosoma 2 (2p21) en 20085. Posteriormente, se descubrió la asociación a mutaciones en el gen SPINT2, aunque con menor frecuencia6.
El trasplante intestinal es hoy en día la única alternativa para estos pacientes con fallo intestinal irreversible3. Nuestros 3 pacientes han sido trasplantados, 2 de ellos han tenido una evolución favorable, presentando en la actualidad autonomía digestiva. El tercero se encuentra pendiente de revaloración terapéutica tras pérdida del injerto por un rechazo exfoliativo.
En conclusión, la DEI es una de las causas de diarrea grave rebelde de comienzo precoz, que produce fracaso intestinal permanente con dependencia indefinida de la NPT. El diagnóstico es clínico, histológico y genético, pero, en ocasiones, puede resultar difícil por su afectación parcheada y variable. El trasplante intestinal es una alternativa terapéutica útil para conseguir la autonomía digestiva y normalizar la calidad de vida de estos pacientes.
