

El síndrome hemofagocítico (SH) constituye una manifestación común a una serie de anomalías congénitas que afectan a la excreción lisosomal, interrumpiendo la vía citolítica gránulodependiente y desencadenando una disfunción de la sinapsis inmunológica. La presencia de manifestaciones características en otros tejidos puede orientar el diagnóstico etiológico.
Pacientes y métodosPresentamos los hallazgos clínicos y biológicos de dos hermanos diagnosticados de linfohistiocitosis hemofagocítica familiar tipo 3 (FHL-3), dos pacientes con síndrome de Griscelli tipo 2 (GS-2), y un síndrome de Chédiak-Higashi (CHS).
ResultadosLos pacientes de FHL-3 aportaron un resultado positivo en el estudio mutacional de UNC13D indicado por un SH precoz en el primero de ellos. El primer diagnóstico de SG-2 se confirmó por la presencia de una mutación en el gen Rab27A en una paciente con SH en la que había un llamativo trastorno de la pigmentación. La misma mutación se detectó en una prima afecta también de trastornos de la pigmentación. El diagnóstico de SCH se realizó en un paciente que presentaba un SH con trastornos de la pigmentación y granulación atípica en células hematopoyéticas. El hallazgo de una mutación en el gen LYST confirmó el diagnóstico.
ConclusionesEn los pacientes con SH primario es preciso atender a manifestaciones extra-inmunológicas características de ciertos trastornos de la secreción lisosomal. La curiosa relación entre albinismo e inmunidad ha jugado recientemente un papel decisivo en la identificación de los mecanismos moleculares involucrados en estos procesos.
Haemophagocytic syndrome (HS) is a common manifestation of several congenital disorders characterised by a disruption of lysosomal secretion, interrupting the cytolytic pathway and triggering a dysfunction in the immune synapse. In this situation, the recognition of certain extra-immunological manifestations may help in the diagnostic process.
Patients and methodsWe describe the clinical and biological features present in two brothers with familial haemophagocytic lymphohistiocytosis type 3 (FHL-3), two patients with Griscelli syndrome type 2 (GS-2) and one patient with Chédiak-Higashi syndrome (CHS).
ResultsMutational assays at UNC13D were carried out on two brothers after diagnosing an early onset HS in the first one, yielding a positive result in both cases with a consequent diagnosis of FHL-3. The diagnosis of GS-2 was supported by positive results of mutational Rab27A studies in one patient with HS and abnormal pigmentation, and in her cousin who was affected by a similar abnormal pigmentation. The diagnosis of CHS was established in one patient with HS, abnormal pigmentation and atypical granules on cytological examination of a bone marrow smear. Diagnosis was confirmed in this patient by the finding of a homozygous LYST mutation.
ConclusionsWe point out the importance of recognising the presence of typical extra-immunological manifestations of certain congenital disorders of lysosome secretion in patients diagnosed with HS. The association of albinism and immunodeficiency has played a critical role in the recent identification of the molecular mechanism involved in these disorders.
El lisosoma es una organela citoplasmática característica de las células eucariotas cuya principal función es la degradación de los productos de desecho intracelular así como del material endocitado. Algunas células especializadas contienen los denominados «orgánulos relacionados con los lisosomas» (ORL), un grupo heterogéneo de vesículas que comparte ciertas características con los lisosomas pero que difieren en cuanto a su función, morfología y composición1,2. Algunos ejemplos son los gránulos líticos de los linfocitos citotóxicos, los melanosomas de los melanocitos o los gránulos delta de las plaquetas. La biogénesis de los ORL emplea una maquinaria común a todos ellos y la heterogeneidad en las manifestaciones clínicas de los diferentes defectos genéticos que la alteran es la consecuencia del tipo de ORL afectado en cada entidad3. El síndrome hemofagocítico (SH) constituye una manifestación común y es debida a la alteración funcional de los ORL involucrados en la vía citolítica gránulodependiente que conduce a una disfunción de la sinapsis inmune4. La disfunción lisosomal en algunas de estas anomalías puede desencadenar manifestaciones clínicas adicionales relacionadas con defectos de secreción de los ORL en otros tejidos y su identificación puede orientar el estudio mutacional permitiendo un diagnóstico molecular precoz.
MétodosPresentamos los hallazgos clínicos y biológicos en 5 pacientes diagnosticados de trastornos de la secreción lisosomal: un paciente afecto de síndrome de Chédiak-Higashi (CHS), dos con síndrome de Griscelli tipo 2 (GS-2) y dos hermanos diagnosticados de linfohistiocitosis hemofagocítica familiar tipo 3 (FHL-3). Los procedimientos diagnósticos y terapéuticos se realizaron previo consentimiento informado de uno de los progenitores.
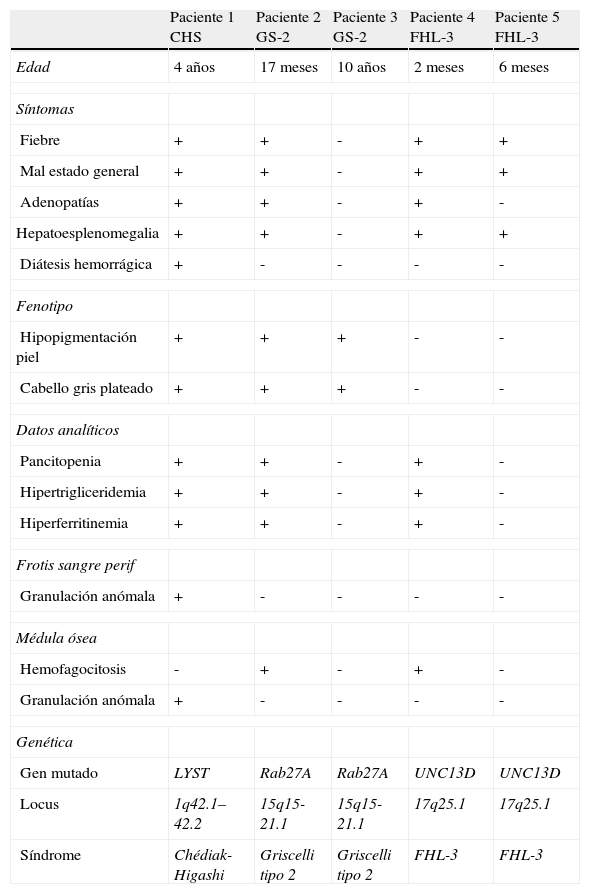
ResultadosEl diagnóstico de CHS se realizó en un paciente afecto de SH con trastornos de la pigmentación (fig. 1a), granulación atípica en células hematopoyéticas (fig. 1b y 1c) y disfunción plaquetaria. El hallazgo de una mutación en el gen LYST confirmó el diagnóstico. El primer diagnóstico de SG-2 se confirmó al comprobar la presencia de una mutación homocigota en Rab27A en una paciente afecta de SH (fig. 1d y 1e), en la que además había un llamativo trastorno de la pigmentación. La misma mutación se detectó en una prima afecta también de trastornos de la pigmentación (fig. 1f), que no había presentado manifestaciones hematológicas. Los dos pacientes de LHH-3 aportaron un resultado positivo en el estudio mutacional de UNC13D indicado por un SH precoz en el primero de ellos. En la tabla 1 se exponen las características clínicas de los pacientes al diagnóstico. Los trastornos de la pigmentación fueron los hallazgos clínicos que determinaron la orientación diagnóstica en los pacientes con SG-2 y CHS.

1a. Paciente 1 afecto de CHS, coloración anómala del cabello. 1b y 1c. Paciente 1 afecto de CHS, granulación intracelular atípica en sangre periférica y médula ósea, respectivamente. 1d. Paciente 2 afecta de SG-2, hepatoesplenomegalia. 1e. Paciente 2 afecta de SG-2, hemofagocitosis en médula ósea. 1f. Paciente 3 afecta de SG-2, coloración anómala de piel y cabello.
Principales características de los pacientes al diagnóstico
| Paciente 1 CHS | Paciente 2 GS-2 | Paciente 3 GS-2 | Paciente 4 FHL-3 | Paciente 5 FHL-3 | |
| Edad | 4años | 17meses | 10años | 2meses | 6meses |
| Síntomas | |||||
| Fiebre | + | + | - | + | + |
| Mal estado general | + | + | - | + | + |
| Adenopatías | + | + | - | + | - |
| Hepatoesplenomegalia | + | + | - | + | + |
| Diátesis hemorrágica | + | - | - | - | - |
| Fenotipo | |||||
| Hipopigmentación piel | + | + | + | - | - |
| Cabello gris plateado | + | + | + | - | - |
| Datos analíticos | |||||
| Pancitopenia | + | + | - | + | - |
| Hipertrigliceridemia | + | + | - | + | - |
| Hiperferritinemia | + | + | - | + | - |
| Frotis sangre perif | |||||
| Granulación anómala | + | - | - | - | - |
| Médula ósea | |||||
| Hemofagocitosis | - | + | - | + | - |
| Granulación anómala | + | - | - | - | - |
| Genética | |||||
| Gen mutado | LYST | Rab27A | Rab27A | UNC13D | UNC13D |
| Locus | 1q42.1–42.2 | 15q15-21.1 | 15q15-21.1 | 17q25.1 | 17q25.1 |
| Síndrome | Chédiak-Higashi | Griscelli tipo 2 | Griscelli tipo 2 | FHL-3 | FHL-3 |
CHS: síndrome de Chédiak-Higashi; FHL-3: linfohistiocitosis hemofagocítica familiar tipo 3; GS-2: síndrome de Griscelli tipo 2.
Las enfermedades genéticas que afectan a la función de los ORL producen diferentes manifestaciones clínicas dependiendo de la estirpe celular afectada.
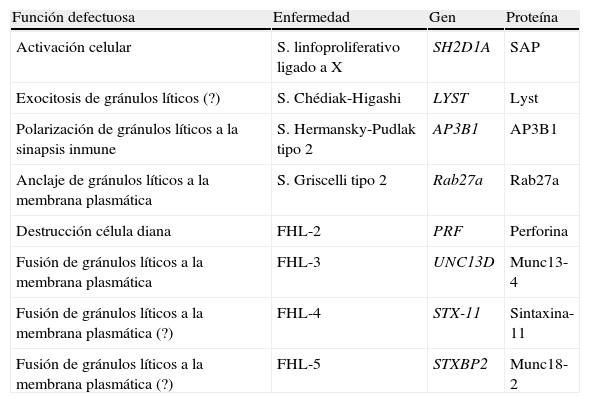
La vía citolítica gránulodependiente requiere una serie de pasos, desde la activación de la célula T, la síntesis de los gránulos líticos, su polarización hacia la sinapsis inmune y la exocitosis de su contenido que una vez liberado ejerce su acción citotóxica sobre la célula diana5,6. El conocimiento de las señales moleculares de este proceso ha permitido describir la patogenia de un grupo de enfermedades que comprometen la actividad citotóxica de estas células (tabla 2) y cuya característica común es el desarrollo de una entidad denominada SH7, caracterizado por infiltración visceral masiva por linfocitos activados y macrófagos con fiebre, hepatoesplenomegalia, citopenias, y menos frecuentemente, infiltración del sistema nervioso central. Otros criterios diagnósticos son la hipofibrinogenemia, hiperferritinemia, hipertrigliceridemia y elevación de los niveles plasmáticos del receptor soluble de IL2 (sCD25) secretado por las células T activadas8. La combinación de quimio e inmunoterapia es capaz de controlar la enfermedad en la mayoría de los casos, aunque rara vez se obtiene una remisión completa y la mayoría de los pacientes fallece de forma prematura a menos que reciba un alotrasplante hematopoyético (TPH). Los pacientes 1 y 2 de nuestra serie debutaron como un SH que respondió bien a tratamiento inmunomodulador sometiéndose posteriormente con éxito a un TPH. Ambos pacientes se encuentran en remisión tras un seguimiento de 24 y 49meses tras el TPH respectivamente. La paciente 3 no ha desarrollado manifestaciones hematológicas y carece de familiar compatible por lo que se adoptó una actitud expectante. Los pacientes 4 y 5 fallecieron a consecuencia de su enfermedad, uno de ellos tras TPH.
Enfermedades genéticas que causan HLH
| Función defectuosa | Enfermedad | Gen | Proteína |
| Activación celular | S. linfoproliferativo ligado a X | SH2D1A | SAP |
| Exocitosis de gránulos líticos (?) | S. Chédiak-Higashi | LYST | Lyst |
| Polarización de gránulos líticos a la sinapsis inmune | S. Hermansky-Pudlak tipo 2 | AP3B1 | AP3B1 |
| Anclaje de gránulos líticos a la membrana plasmática | S. Griscelli tipo 2 | Rab27a | Rab27a |
| Destrucción célula diana | FHL-2 | PRF | Perforina |
| Fusión de gránulos líticos a la membrana plasmática | FHL-3 | UNC13D | Munc13-4 |
| Fusión de gránulos líticos a la membrana plasmática (?) | FHL-4 | STX-11 | Sintaxina-11 |
| Fusión de gránulos líticos a la membrana plasmática (?) | FHL-5 | STXBP2 | Munc18-2 |
FHL-2: linfohistiocitosis hemofagocítica familiar tipo 2; FHL-5: linfohistiocitosis hemofagocítica familiar tipo 5; FHL-4: linfohistiocitosis hemofagocítica familiar tipo 4; FHL-3: linfohistiocitosis hemofagocítica familiar tipo 3.
(?) Mecanismo no aclarado10,16–18
La pigmentación normal de piel y cabello requiere la transferencia de los melanosomas desde los melanocitos a los queratinocitos adyacentes. La presencia de alteraciones de la pigmentación y trastornos inmunes en los síndromes de GS-2, CHS y Hermansky-Pudlak tipo 2 sugirió que los melanocitos y las células del sistema inmune podrían utilizar una vía secretora común9. La identificación de los melanosomas como ORL involucrados en la secreción de melanina ha permitido dar explicación a la patogenia estos trastornos. En nuestra serie el trastorno de la pigmentación resultó determinante para orientar los diagnósticos de CHS (paciente 1) y SG-2 (paciente 2 y 3).
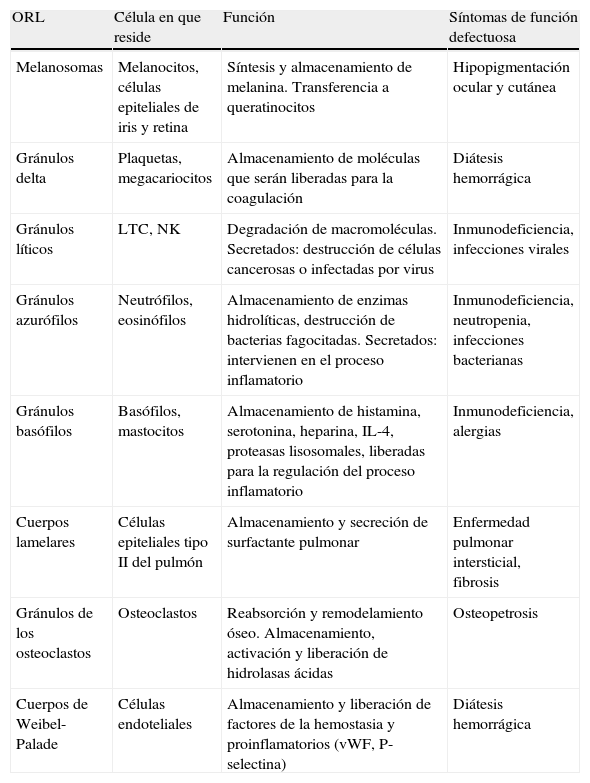
En algunas de estas anomalías, la disfunción de los ORL en otros tejidos provoca además otras manifestaciones cuya identificación puede orientar el diagnóstico. En la tabla 3 se exponen la función, el tipo de célula y la relevancia clínica de los principales ORL.
Principales ORL y sus funciones
| ORL | Célula en que reside | Función | Síntomas de función defectuosa |
| Melanosomas | Melanocitos, células epiteliales de iris y retina | Síntesis y almacenamiento de melanina. Transferencia a queratinocitos | Hipopigmentación ocular y cutánea |
| Gránulos delta | Plaquetas, megacariocitos | Almacenamiento de moléculas que serán liberadas para la coagulación | Diátesis hemorrágica |
| Gránulos líticos | LTC, NK | Degradación de macromoléculas. Secretados: destrucción de células cancerosas o infectadas por virus | Inmunodeficiencia, infecciones virales |
| Gránulos azurófilos | Neutrófilos, eosinófilos | Almacenamiento de enzimas hidrolíticas, destrucción de bacterias fagocitadas. Secretados: intervienen en el proceso inflamatorio | Inmunodeficiencia, neutropenia, infecciones bacterianas |
| Gránulos basófilos | Basófilos, mastocitos | Almacenamiento de histamina, serotonina, heparina, IL-4, proteasas lisosomales, liberadas para la regulación del proceso inflamatorio | Inmunodeficiencia, alergias |
| Cuerpos lamelares | Células epiteliales tipo II del pulmón | Almacenamiento y secreción de surfactante pulmonar | Enfermedad pulmonar intersticial, fibrosis |
| Gránulos de los osteoclastos | Osteoclastos | Reabsorción y remodelamiento óseo. Almacenamiento, activación y liberación de hidrolasas ácidas | Osteopetrosis |
| Cuerpos de Weibel-Palade | Células endoteliales | Almacenamiento y liberación de factores de la hemostasia y proinflamatorios (vWF, P-selectina) | Diátesis hemorrágica |
ORL: orgánulos relacionados con los lisosomas; vWF: factor de von Willebrand.
El SCH es debido a una mutación en el gen LYST que origina una proteína anómala responsable, por mecanismos aún no totalmente descritos, de la formación de unos gránulos gigantes incapaces de fusionarse con la membrana plasmática10. Este fenómeno repercute en diversas líneas celulares lo que explica las manifestaciones clínicas de esta enfermedad. La actividad citotóxica de los linfocitos T-CD8+ está alterada, por la incapacidad de los gránulos líticos para liberar su contenido. El albinismo oculocutáneo y la coloración gris metalizada del cabello son característicos y una anomalía en los melanosomas que les impide ser transferidos adecuadamente a los queratinocitos. En nuestro paciente 1, el albinismo y la presencia de gránulos anómalos en las células hematopoyéticas resultaron determinantes para el diagnóstico. A pesar de un recuento plaquetario normal, el tiempo de hemorragia está prolongado por alteración de la agregación plaquetaria, debida a un déficit de la secreción de los gránulos delta11. La neutropenia característica confiere una mayor predisposición a las infecciones piógenas y es debida a la agregación espontánea de moléculas de superficie de los megagránulos anómalos y de los gránulos secundarios normales que produce activación celular y finalmente apoptosis12.
El SG-2 es una enfermedad autosómica recesiva debida a la mutación del gen Rab27A que impide el anclaje de los gránulos secretores a la membrana plasmática, paso previo a la fusión entre ambos. Al igual que en el SCH, estos pacientes presentan anomalías en la pigmentación cutánea y del cabello pero no presentan la típica granulación citoplasmática característica del SCH. Asimismo, la alteración de la función inmune reside en la incapacidad de los linfocitos T-CD8+ para liberar el contenido de sus gránulos líticos y es la base, al igual que en el SCH, para el desarrollo de la fase acelerada de la enfermedad13,14.
En la FHL-3, los gránulos secretores de los linfocitos son capaces de anclarse a la membrana plasmática, pero el defecto reside en su imposibilidad para fusionarse con ella. La mutación se sitúa en el gen UNC13D y solo afecta a una línea celular, por lo que no se acompaña de manifestaciones extrainmunológicas15.
En conclusión, los trastornos de la secreción lisosomal son un grupo de anomalías en las señales moleculares que impiden la correcta migración y secreción del contenido de los lisosomas y ORL. En las células del sistema inmune, se produce una interrupción de la vía citolítica gránulo-dependiente, y consecuentemente, un trastorno de la inmunidad celular. El SH es la denominada «fase acelerada» de la enfermedad, caracterizado por una proliferación linfohistiocítica incontrolada, con afectación del estado general y fallo multiorgánico. En algunas de estas anomalías, la disfunción lisosomal y ORL en otros tejidos provoca otras manifestaciones que pueden orientar el estudio mutacional.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Nuestro agradecimiento a Karim Beutel y Udo Zur Stadt (Department of Pediatric Hematology and Oncology, Children's Hospital, University of Hamburg, Alemania) y a Marisa Lozano, Jose Rivera y Francisco Ortuño (Centro de Hemodonación, Servicio Murciano de Salud) por la realización de los estudios moleculares.
