



La intolerancia hereditaria a la fructosa es un error congénito del metabolismo de herencia autosómica recesiva, secundaria a la deficiencia o ausencia de la enzima aldolasa B que ocasiona el acúmulo de fructosa 1-fosfato. Su incidencia es aproximadamente de 1:20.000 y la sintomatología comienza tras la introducción de la fruta, por la cual sienten gran aversión1,2. La ingesta aguda de fructosa ocasiona vómitos, sudoración, letargia y/o convulsiones, acompañándose generalmente de hipoglucemia. El consumo crónico, a expensas de ingesta diaria baja, provoca retraso del crecimiento, hepatopatía, nefropatía y vómitos intermitentes3. Estas manifestaciones se intensifican con enfermedades infecciosas intercurrentes, como en el caso que presentamos.
Se trata de una lactante de 17 meses que acude al servicio de Urgencias por vómitos, febrícula y rechazo de la ingesta en los últimos 10 días, habiendo recibido cefixima ante la sospecha de infección urinaria.
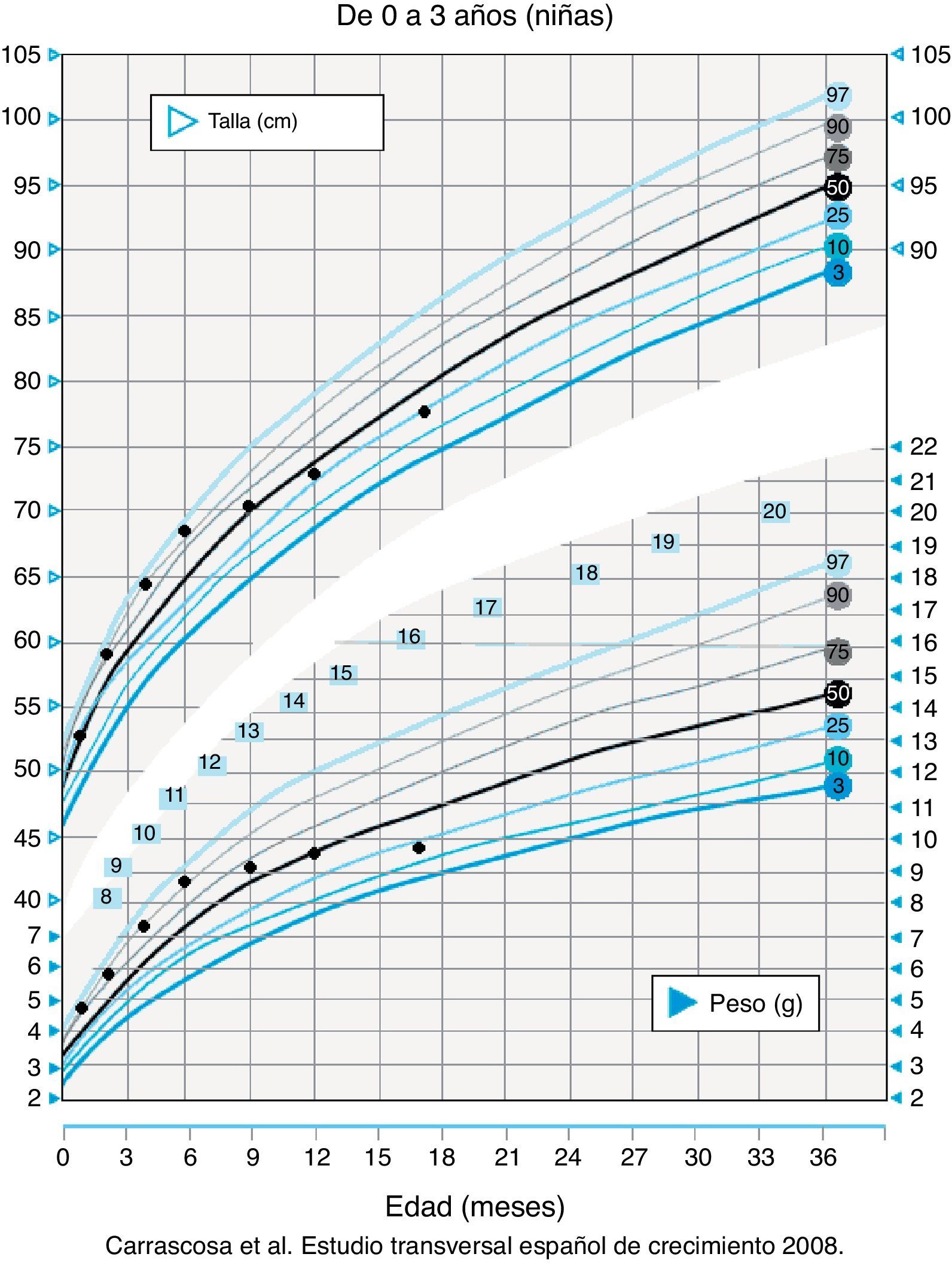
No presentó antecedentes perinatales de interés, siendo el cribado metabólico y el desarrollo psicomotor normales. Recibió lactancia artificial exclusiva durante los primeros 6 meses con posterior introducción del beikost sin incidencias, salvo inicialmente presencia de vómitos con la fruta, que en el momento del ingreso ingiere en escasa cantidad. Presentó adecuada ganancia ponderoestatural hasta el séptimo mes, en el que inició un moderado retraso del crecimiento (fig. 1). No existen consanguinidad familiar ni antecedentes familiares de interés. Convive con un gato en el domicilio.
En la exploración física, destacaba palidez, sequedad mucocutánea, hepatomegalia (3 traveses) y esplenomegalia (un través). La gasometría capilar mostraba acidosis metabólica compensada y la bioquímica sanguínea evidenciaban hipertransaminasemia (GPT 328 UI/L, GOT 470 UI/L), hiperbilirrubinemia de predominio directo (3,3mg/dL), hipouricemia y normoglucemia. Ingresó, se instauró fluidoterapia y a las 24h alimentación adaptada, apreciándose buena evolución clínica y normalización de valores de transaminasas a los 12 días.
Un estudio analítico más completo evidenció hipertrigliceridemia, hipercolesterolemia y discreto alargamiento del tiempo de protrombina, así como microalbuminuria, glucosuria y excreción del ácido úrico elevado en orina de 24h. La ecografía abdominal mostraba un ligero aumento del bazo y un hígado aumentado de tamaño, con ecogenicidad normal sin infiltración hepatocelular ni obstrucción biliar.
Inicialmente, se descartó etiología infecciosa viral (serologías negativas de virus hepatotropos), tuberculosis e infección por parásitos, incluyendo determinación de Leishmania en orina, serología de Bartonella quintana y henselae, estudio de Toxoplasma, Plasmodium y microfilarias, que fueron negativos. No se hallaron sustancias tóxicas en orina y se excluyeron causas autoinmunes (anticuerpo antinuclear, anticuerpo antimúsculo liso, antimitocondriales y LKM: negativos).
A los 45 días de ser dada de alta, ingresa nuevamente con hallazgos analíticos similares (GPT 1.055 UI/L, GOT 2.091 UI/L) y la misma sintomatología, esta vez acompañada de fiebre alta, tos y rinorrea. Se completó el estudio descartando enfermedad celiaca, proceso linfoproliferativo, enfermedad de Wilson, déficit de alfa-1-antitripsina y fibrosis quística. Asimismo, se realizó detección sistemática de errores congénitos de metabolismo, con cribado de azúcares en orina normal. En el momento agudo del cuadro, se detectó elevación de transferrina deficiente en hidratos de carbono (CDT 19%). Ante la sospecha de intolerancia hereditaria a la fructosa, se solicitó un estudio genético que evidenció la mutación (c.360_363delCAAA) del gen de la aldolasa B en homocigosis. Se estableció una dieta exenta de fructosa, galactosa y sorbitol, así como la no utilización de productos de higiene o farmacológicos que contuviesen dichos azúcares, objetivándose a los 23 meses de edad una adecuada ganancia ponderal y normalización de los valores de CDT (1,64%).
En aquellos pacientes con intolerancia hereditaria a la fructosa, que realizan una baja ingesta diaria de la misma, se produce afectación hepática, consistente en hipertransaminasemia, hiperbilirrubinemia, hepatomegalia y alteraciones de la coagulación, junto con afectación tubular proximal que se manifiesta con microalbuminuria, hiperaminoaciduria, glucosuria y excreción elevada de ácido úrico1,2, todos ellos presentes en el caso descrito.
La sospecha diagnóstica obliga a la detección de sustancias reductoras en orina (no presentes en nuestra paciente) y de elevación de CDT, secundaria al acúmulo de fructosa-1-fosfato, que actúa como inhibidor competitivo de la enzima fosfomanomutasa, produciendo un defecto de glucosilación de la transferrina sérica4. Asimismo, la determinación de CDT resulta clave en el seguimiento de estos pacientes, ya que detectaría la realización de una dieta inadecuada5,6. Finalmente, el diagnóstico de certeza se obtiene al detectar mutaciones del gen de la aldolasa B3,7 o demostrando el déficit enzimático en muestras de tejido afectado.
Aunque la presencia de fiebre en pacientes con hipertransaminasemia, como ocurría en el caso descrito, debe hacernos descartar en primer lugar etiologías más frecuentes, como infecciones por virus hepatotropos y enfermedades autoinmunes8, no debemos olvidarnos de la descompensación de las enfermedades metabólicas en el contexto de un proceso intercurrente febril.
