


La respuesta inmunitaria incluye una compleja red de mecanismos de defensa que está formada por medios de barrera (epitelio bronquial), componentes celulares y mediadores solubles. La respuesta inmunitaria normal tiene dos brazos de actuación: un sistema inespecífico de acción rápida frente al inicio de la infección y un sistema específico inmunitario, organismo selectivo, más tardío. El sistema inespecífico está formado por células fagocíticas (neutrófilos y macrófagos), células natural killer (NK) y células presentadoras de antígenos que iniciarán la respuesta específica. Las proteínas del complemento promueven la inflamación inicial y facilitan la muerte de organismos extracelulares. El sistema específico esta formado por los linfocitos T y B, responsables de la inmunidad celular y humoral, respectivamente. La inmunidad celular interviene en la defensa frente a organismos intracelulares como virus, parásitos y micobacterias. Los linfocitos B son responsables de la inmunidad humoral; a través de la formación de anticuerpos dificultan la diseminación de patógenos extracelulares. Intervienen en la defensa frente a bacterias encapsuladas como el neumococo. La respuesta humoral y celular no son independientes y un funcionamiento alterado de un tipo de respuesta puede influir en el otro componente1.
Las inmunodeficiencias primarias son enfermedades hereditarias que afectan al sistema inmunitario. Pueden deberse a la alteración de un solo gen, ser poligénicas o pueden representar la interacción de determinadas características genéticas y factores ambientales o infecciosos2. Representan un grupo heterogéneo que se caracteriza por la predisposición a enfermedades infecciosas, autoinmunitarias y procesos cancerosos. La prevalencia de las inmunodeficiencias primarias en los diferentes países varía dependiendo de los procedimientos técnicos empleados, de las clasificaciones utilizadas y de la inclusión o no de pequeños defectos inmunitarios. En países pertenecientes al registro europeo de inmunodeficiencias como Noruega, la tasa es de 6,82 por 100.000 habitantes3. Países como Australia que no incluyen déficit de inmunoglobulina A (IgA) o de producción de anticuerpos asintomáticos ni déficit de complemento, las tasas bajan a 2,82 por 100.000 habitantes4. La distribución de los de los déficit inmunitarios varía según los criterios de inclusión. En el registro español 46,5 % son déficit de IgA, 25,1 % inmunodeficiencia común variable, 7,1 % inmunodeficiencia severa combinada, 6,2 % déficit de C1 inhibidor, 5,8 % agammaglobulinemia ligada al cromosoma X, 5,6 % déficit de subclases de IgG y 3,7 % enfermedad granulomatosa crónica5. En todas las series el 50-60 % del total de las inmunodeficiencias son defectos de la inmunidad humoral que además son las que dan origen a manifestaciones fundamentalmente respiratorias6. Este grupo de inmunodeficiencias es el más frecuente tanto en la edad adulta como en el grupo pediátrico. Por sexos las inmunodeficiencias son más frecuentes en varones con una relación 2:1, por el peso de las inmunodeficiencias ligadas al cromosoma X, predominio que aumenta a 3:1 en los niños muertos por inmunodeficiencias graves3. Las inmunodeficiencias, sobre todo las menos severas, se van a manifestar clínicamente con síntomas respiratorios y por ello forman parte del diagnóstico diferencial de un niño con problemas respiratorios7-12.
Manifestaciones pulmonares de las inmunodeficiencias
La sintomatología respiratoria es la primera manifestación de una inmunodeficiencia primaria en el 70,3 % de las ocasiones y el 61,3 % de los niños con inmunodeficiencia precisan en alguna ocasión asistencia en una consulta de respiratorio infantil en un hospital terciario de nuestro medio10. Las manifestaciones pulmonares de las inmunodeficiencias primarias las podríamos clasificar en grupos: neumonías por gérmenes no habituales, infecciones respiratorias sinopulmonares de repetición y neumonías de evolución tórpida.
Neumonías por gérmenes no habituales
Pneumocystis carinii es un patógeno oportunista frecuente en niños con una función anormal de los linfocitos T, bien primaria o secundaria12. Una neumonía por P. carinii es frecuentemente la primera manifestación clínica de una inmunodeficiencia severa combinada. En la serie de Berrington y Flood7, 10 de 50 niños transferidos a una unidad suprarregional de trasplantes de médula ósea tenían una neumonía por P. carinii, diagnosticada por lavado broncoalveolar, en el momento de su ingreso. Solamente en uno de ellos se había sospechado su diagnóstico antes del traslado, a pesar de tener todos ellos sintomatología respiratoria. La edad media de los niños fue de 6,5 meses con un rango de 4,5 meses y un año. Todos ellos presentaban inmunodeficiencias severas, 5 eran inmunodeficiencias severas combinadas y 2 síndromes de Omenn. La posibilidad de una inmunodeficiencia y una infección por P. carinii debería estar siempre presente en el diagnóstico de un niño con sintomatología respiratoria que se acompaña de mala ganancia ponderal, diarrea crónica o candidiasis oral de repetición7. Por otra parte, debería excluirse una inmunodeficiencia primaria o secundaria en todo lactante con una neumonía por P. carinii.
Infecciones respiratorias de repetición
Llegados a este punto, la primera cuestión es cuántas infecciones puede tener un niño normal. Se considera anormal en un niño una cifra igual o superior a ocho otitis o dos neumonías, sinusitis severas o infecciones sistémicas13,14. Las inmunodeficiencias son una causa infrecuente de infecciones de repetición en el niño8,15. Causas más frecuentes son el asma, bronquitis eosinófila y el llamado "niño normal pero con mala suerte" por Rubin16. Estos niños tienen un crecimiento normal, no tienen antecedentes familiares de defectos de inmunidad, carecen de historia de infecciones de repetición de otra localización y la radiografía de tórax es normal entre los episodios. El pronóstico es bueno. Son niños en el límite superior de la distribución normal del número de infecciones respiratorias8. La posibilidad de una inmunodeficiencia debe sospecharse si las infecciones son especialmente severas y recurrentes, no se controlan con tratamientos convencionales y están acompañadas de retraso en el crecimiento o eccema severo. En estos pacientes el diagnóstico es por desgracia muchas veces tardío y presentan ya alteraciones irreversibles pulmonares, manifestaciones que se correlacionan positivamente con la tardanza del diagnóstico9,17.
Neumonías de evolución tórpida
Aproximadamente el 76 % de las enfermedades granulomatosas crónicas con diagnosticadas antes de los 5 años18 y una gran mayoría lo son antes de los 2 años19,20. La enfermedad granulomatosa crónica es una enfermedad hereditaria recesiva ligada al cromosoma X y ocasionalmente autosómico recesiva18. Estos pacientes tienen uno o varios defectos moleculares del sistema nicotinamida adenindinucleótido fosfato reducido (NADPH) oxidasa de las células fagocíticas que conlleva una fagocitosis anormal de gérmenes catalasa positivos21,22. El 80 % de los pacientes presentan neumonía, siendo el germen más habitual Aspergillus (41 %). Otros gérmenes causantes de neumonía en estos pacientes son: estafilococo (11 %), Burkholderia cepacia (7 %) y Nocardia (6 %)18. Estos niños tienen neumonías de evolución tórpida, o abcesificadas (botriomicosis)23-30. El diagnóstico de enfermedad granulomatosa crónica debería tenerse siempre en cuenta, especialmente si hay historia de abscesos e infecciones de otra localización18. Otros defectos inmunitarios de la inmunidad humoral y celular se manifiestan también como neumonías de evolución tórpida8,9,16,31,32.
Diagnosticando a un niño con sospecha de inmunodeficiencia primaria
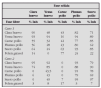
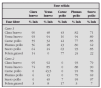
Las inmunodeficiencias son entidades poco frecuentes5. En niños con infecciones respiratorias de repetición, es obligatorio descartar primero entidades de mucha mayor prevalencia en este grupo de edad, como la rinitis alérgica, asma o fibrosis quística de páncreas31. Un lactante con una neumonía por P. carinii, es más probable que padezca de un síndrome de inmunodeficiencia adquirida. Un niño con una neumonía de evolución tórpida o neumonías recurrentes de la misma localización, tendrá como causa más frecuente de su patología respiratoria un cuerpo extraño que una enfermedad granulomatosa crónica, cuya incidencia poblacional es solamente de 1/200.000 a 250.00018. Hay signos de alarma que deberían hacer pensar en una inmunodeficiencia primaria (tabla 1)1,33.

En un niño con algún dato de alarma de inmunodeficiencia (fig. 1), dado que las inmunodeficiencias son enfermedades hereditarias, la primera cuestión a preguntarse es si existe una historia familiar de inmunodeficiencias y, particularmente, si hay varones afectados en la familia (la herencia más frecuente es recesiva ligada al sexo)3. Si no existe historia familiar, teniendo en cuenta que las inmunodeficiencias primarias son poco frecuentes, debemos preguntarnos si la sintomatología puede ser secundaria a otras causas: asma, rinitis alérgica, síndrome de inmunodeficiencia adquirida, malformación pulmonar, cuerpo extraño, etc. Si existe historia familiar o se han descartado estas enfermedades, la edad de inicio orienta en la clase de inmunodeficiencia que puede padecer. Si la clínica se ha iniciado antes de los 6 meses, puede tratarse de una inmunodeficiencia severa que afecta a la inmunidad humoral y celular. Las inmunodeficiencias humorales se inician después de los 6 meses, coincidiendo con la bajada de anticuerpos transferidos por la madre6.

Figura 1. Evaluación de la inmunidad en una consulta de respiratorio. VIH, virus de la inmunodeficiencia humana.
El siguiente paso sería hacer un detección inicial de inmunodeficiencias que incluiría una hematimetría completa con recuento manual, determinación de inmunoglobulinas y tal vez recuento de linfocitos, si es posible en nuestro centro, comparando con las cifras de normalidad para la edad34,35: CD3+ (linfocitos T totales), CD4+/CD3+ (T helper), CD8+/CD3+ (T citotóxico), CD19+ (linfocitos B) CD16+ o CD56+ (células NK). Dependiendo del grupo sanguíneo pueden hacerse isohemaglutininas que en 70-80 % de los casos están presentes para el año de edad. La ausencia de anticuerpos IgM clase específico frente al grupo sanguíneo, implicaría una pobre síntesis de IgM13.
Si nuestro laboratorio lo permite podríamos determinar anticuerpos antitétanos (subclases IgG1 e IgG3) y testar así la producción de anticuerpos frente a antígeno proteico. Se puede investigar también la presencia de IgG antirrubéola en lactantes vacunados. La inexistencia de anticuerpos en niños vacunados también orientaría en una deficiente formación de anticuerpos6. Si todo ello es normal, estudiaríamos la formación de Ac frente a antígeno capsular (subclases IgG2 e IgG4) a las 4 semanas de vacunación con la vacuna polisacárida de Haemophylus o neumococo que tiene más valor en niños mayores de 2 años (los lactantes pueden tener formación anormal de anticuerpos con la vacuna polisacárida)36-43. Se considera una respuesta positiva un título cuatro veces mayor que el valor preinmunización o un valor mayor que 1,3 μg/ml (equivalente a 200 ng AbN/ml)42. En un metaanálisis se cuestiona este límite para el diagnóstico de una inmunodeficiencia. En algunas series, algunos individuos normales, incluso no duplican el título prevacunación en alguno de los serotipos44. El componente de antígeno de pared celular en la vacuna y su presencia en los reactivos puede dar origen a títulos falsamente elevados, en la determinación por radioinmunoanálisis (RIA) o análisis de inmunoabsorción ligado a enzimas (ELISA). Otros contaminantes como antígenos de grupo sanguíneo tienen estructuras antigénicas similares a alguno de los determinantes antigénicos polisacáridos45. Otra variabilidad añadida proviene de cómo se cuantifique la respuesta: IgG específica total o individual para cada serotipo y de la edad de los pacientes, los niños pequeños tienen una respuesta pobre con la vacuna polisacárida44. El problema de la edad podría soslayarse con la vacuna conjugada, pero la presencia de componentes proteicos en la vacuna conjugada invalidan su uso para la valoración de la respuesta ante antígeno capsular por ser más una respuesta IgG1 que IgG231.
Es cuestionable el valor de las subclases de IgG en el diagnóstico de una inmunodeficiencia humoral. Un nivel bajo de IgG1 se observa en inmunodeficiencias pero nunca como fenómeno aislado. Un nivel bajo de IgG2 suele asociarse a infecciones sinopulmonares de repetición pero hay pacientes asintomáticos. Defectos aislados de las subclases 3 y 4 no se asocian a inmunodeficiencias. Si a esto añadimos variabilidades interlaboratorio de hasta el 62 % en la determinación de algunas subclases, parece mucho más lógico determinar la formación de anticuerpos después de la vacunación46.
La valoración básica de la inmunidad celular se encuentra realizada con el recuento de linfocitos T. En individuos normales la cifra de linfocitos T es el 50-80 % de la cifra total de linfocitos con un recuento total de al menos 1.000/mm3. Se determinan también poblaciones de CD4 y CD8 que deben de tener una relación aproximada de 1,5 a 2. Si no se dispone de esta técnica puede realizarse una prueba cutánea con varios antígenos: Candida, tétanos, parotiditis, tuberculina, etc. Existe un multitex comercializado, Multitex CMI®, de eficacia no establecida en niños. Un resultado normal prácticamente excluye un defecto de inmunidad celular. Un resultado negativo en lactantes no es diagnóstico de defecto de inmunidad celular por la posibilidad de una sensibilización insuficiente previa6.
El análisis de la función fagocitaria puede hacerse con un recuento de neutrófilos y un test de nitroazul de tetrazolio (NBT)6.
Un resultado anormal en alguna de estas determinaciones o la fundamentada sospecha de una inmunodeficiencia, a pesar de la normalidad de este nivel básico de detección, hace aconsejable la consulta con un inmunólogo1.
Conclusiones
Las inmunodeficiencias primarias forman parte del diagnóstico diferencial de un niño con neumonías y procesos respiratorios de repetición, neumonías de evolución tórpida y neumonías con gérmenes no habituales. A pesar de que se han descrito más de 100 defectos inmunitarios diferentes, la inmensa mayoría son déficit de inmunidad humoral que pueden ser confirmados con la historia clínica y unas pruebas de laboratorio básicas. La complejidad de su diagnóstico definitivo (en más de 75 existe diagnóstico genético molecular) y de su tratamiento, hace obligatorio la consulta con un inmunólogo.
