


Sr. Editor:
La hipocondroplasia (OMIM 146000) es una osteocondrodisplasia, no infrecuente, que se hereda con carácter autosómico dominante, aunque se detectan numerosos casos esporádicos 1-5.
La hipocondroplasia no suele manifestarse hasta la niñez, cuando se descubre una talla baja desproporcionada con miembros cortos, se afectan los segmentos proximales (rizomélicos). Los niños tienen una complexión fornida, ligera prominencia frontal, braquidactilia leve o moderada, manos y pies toscos, el tamaño de la cabeza es normal generalmente y la lordosis lumbar está aumentada. El dato clínico más distintivo es la ausencia del estirón puberal de crecimiento. El arqueamiento de las piernas se desarrolla durante la niñez temprana pero, a menudo, mejora espontáneamente. Radiológicamente los huesos largos son cortos y gruesos, los ilíacos son más pequeños de lo normal, las cabezas femorales son cortas y anchas, el peroné distal es más largo que la tibia distal. La distancia interpedicular se estrecha desde L1 hasta L5 y los pedículos son cortos. Las complicaciones son raras y algunos casos nunca son diagnosticados 3-5.
Se trata de una enfermedad genética que sucede por mutaciones, en el dominio proximal tirosincinasa, del gen que codifica el receptor 3 del factor de crecimiento fibroblástico (FGFR3), localizado en el cromosoma 4 (4p16.3).
Otras mutaciones en el gen FGFR3 son también responsables de la acondroplasia, la displasia tanatofórica tipo I y la displasia tanatofórica tipo II (grupo acondroplasia) 1,2. Se trata de un niño de 10 años y 3 meses de edad que presenta talla baja desproporcionada y fue diagnosticado de hipocondroplasia, por criterios clínicos-radiológicos, a los 2 años de edad.
Antecedentes personales: embarazo controlado sin patología, a término; parto eutócico; al nacimiento: longitud 49 cm (0,60 DE), peso 3.800 g (0,64 DE), perímetro craneal 35,5 cm (0,53 DE); desarrollo psicomotor normal. La talla, a partir de los 2 años, ha oscilado entre 3 y 4 DE en relación a una población de niños normales 6 y de forma paralela y discretamente superior a la curva de distancia para niños afectos de hipocondroplasia 7. La velocidad de crecimiento (VC) se ha modificado entre 0 y 1 DE durante el período 2 a 8 años de edad y entre 1 y 2 DE en el período 8 a 10 años.
No antecedentes familiares de talla baja, retraso puberal ni dismorfias. Padres sanos no consanguíneos. Talla materna 165,5 cm (0,74 DE). Talla paterna 175 cm (0,09 DE). Talla diana o genética 176,7 cm (0,18 DE).
Características fenotípicas: talla baja, ligera prominencia frontal, braquicefalia, región occipital plana, micromelia (con estructura normal), manos y pies cortos, braquidactilia.
Valoración auxológica y madurativa: talla, 119,2 cm (3,5 DE); relación segmento superior/segmento inferior (SS/SI), 1,4; peso, 24,7 kg (1,51 DE); VC, 3,9 cm/año (1,01 DE); volumen testicular 3 ml.
Determinaciones bioquímicas: hemograma, VSG, bioquímica general, sedimento de orina, estudio metabólico del hierro, perfil celíaco: normales.
Determinaciones hormonales plasmáticas: TSH, T4 libre, anti-TPO normales; IGF-I (IRMA no extractivo): 28,00 nMol/l (rango, 7,00-68,00), IGF-BP3 (IRMA): 4.133,00 ng/ml (rango, 1.250,00-7.060,00); test de clonidina para GH (RIA): GH basal 0,20 ng/ml, GH pico: 4,4 ng/ml; test de hipoglucemia insulínica para GH: GH basal 0,20 ng/ml, GH pico 2,20 ng/ml.
Estudios de imagen:
1. Radiografía de mano y muñeca izquierda: edad ósea, 9 años (Greulich y Pyle); relación edad ósea/edad cronológica 9/10,25 = 0,87.
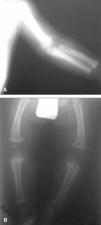
2. Mapas óseos (edad 16 meses): acortamiento de miembros, disminución de longitud de fémures y húmeros con metáfisis anchas y toscas. Disminución de la distancia interpedicular de las últimas vértebras lumbares. Hipoplasia del tercio inferior de ambos ilíacos con horizontalización de los acetábulos. Ensanchamiento de arcos costales (fig. 1).
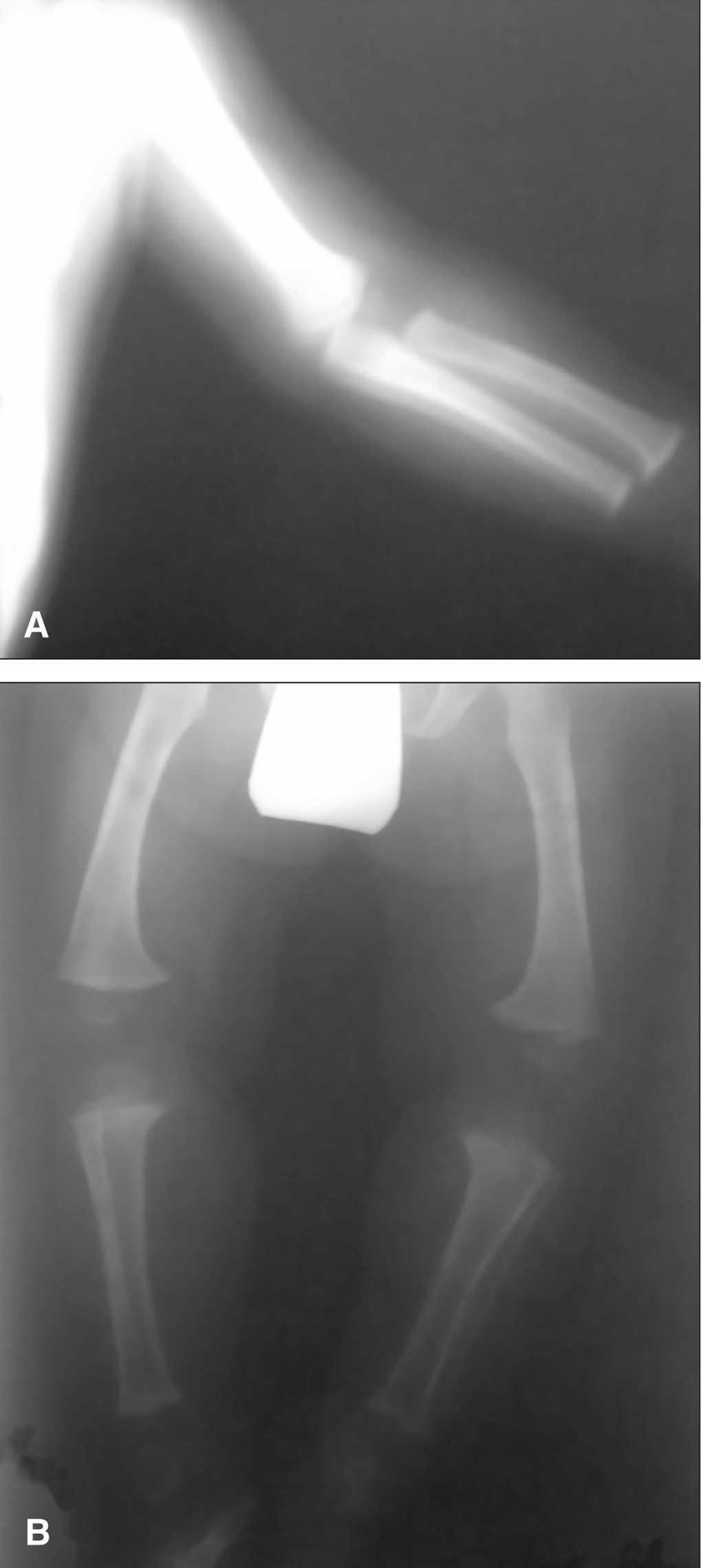
Figura 1. Acortamiento de miembro superiorA)e inferioresB), con disminución de longitud de fémures y húmero, con metáfisis anchas y toscas.
3. Mapa óseo (10 años de edad): también se aprecia disminución de la densidad ósea en manos y muñecas. Pelvis de aspecto "cuadrado".
4. Estudio molecular para hipocondroplasia (técnica de reacción en cadena de polimerasa): mutación N540K.
El paciente presenta una talla baja (3,5 DE) desproporcionada con miembros cortos (relación SS/SI aumentada) desde la primera infancia y la velocidad de crecimiento es normal hasta la edad de 8 años, momento en que se produce una desaceleración de la misma. La relación EO/EC es 0,87 a los 10 años.
Las características clínicas y el mapa óseo son indicativos de hipocondropasia.
Según un estudio realizado por Prinster et al 8 las regiones esqueléticas cruciales en el diagnóstico clínico-radiológico de hipocondroplasia son la zona lumbar y las piernas mientras que la pelvis y las manos son menos características; de ahí que sea necesario, para reducir el riesgo de falsos diagnósticos, hacer una evaluación cuidadosa de ambas regiones, sobre todo en los casos sin alteración genética definida.
En la hipocondroplasia la talla es baja desde la niñez y la velocidad de crecimiento resulta, generalmente, normal, en la primera década hasta la pubertad en la cual no se produce el estirón de crecimiento, lo que disminuye significativamente la talla adulta de estos pacientes (145-165 cm en niños, 133,4-150,6 cm en niñas) 3. En el caso comentado se constató talla baja desproporcionada desde la primera infancia, en clara discordancia con la talla diana o genética (0,18 DE). También, un descenso prepuberal en la velocidad de crecimiento a partir de los 8 años de edad, propio de la hipocondroplasia.
Los valores de IGF-1 e IGF-BP3 se hallan dentro de un rango normal para la edad, aunque los test farmacológicos de estímulo para GH mostraron una respuesta baja. En los niños con talla baja y valores de IGF-1 e IGF-BP3 normales es importante descartar una displasia ósea ya que los test para estimulación de GH, en fase prepuberal y precoz de la pubertad, pueden tener un alto índice de falsos positivos 9.
El diagnóstico de hipocondroplasia basado en hallazgos clínicos y radiológicos es, a menudo, incierto. Por técnicas de genética molecular (PCR) se estudia el gen FGFR3 que codifica el receptor 3 del factor de crecimiento fibroblástico (FGFR3), localizado en el cromosoma 4 (4p16.3). La mutación principal, mutación N540K, resulta en una sustitución Asn540Lys. No obstante, la hipocondroplasia es un trastorno genéticamente heterogéneo que puede estar causado por mutaciones en genes diferentes al FGFR3 o, quizá, por mutaciones no detectadas en este gen 10.
Según un estudio realizado por Grigelioniene et al 10, individuos con hipocondroplasia afectados de la mutación Asn540Lys, como el caso comentado, presentan mayor desproporción de segmentos corporales que el resto.
Consideramos que todos los pacientes con diagnóstico de acondroplasia o hipocondroplasia deben ser estudiados molecularmente. La importancia de un diagnóstico clínico y su confirmación molecular radica en un correcto consejo genético para el paciente y su familia así también como un asesoramiento sobre la evolución de talla y el desarrollo general del mismo.
Correspondencia: Dra. L. Bardallo Cruzado.
Avda. Parque de Doñana, 16, 6.º A. 41015 Sevilla. España.
Correo electrónico: berja@us.es
