



Introducción
El síndrome de Shwachman-Diamond (SDS) es la segunda causa más frecuente de insuficiencia pancreática exocrina, después de la fibrosis quística. Fue descrito por primera vez en 1964 por Shwachman, Diamond, Oski y Khaw en Estados Unidos 1 y Bodia, Sheldon y Lightwood en Gran Bretaña 2. Observaron un grupo de pacientes con fallo de crecimiento en la infancia e insuficiencia pancreática exocrina con diarrea y anormalidades hematológicas. Realizaron biopsias pancreáticas en pacientes seleccionados revelando un reemplazamiento de tejido adiposo de los ácinos con preservación de los islotes de Langerhans. La médula ósea fue hipoplásica con fibrosis y grasa aumentada; muchos pacientes tenían hepatomegalia. Tres años más tarde Burke et al 3 estudiaron una serie de 19 pacientes con SDS en Australia; aproximadamente la mitad tenían malabsorción y esteatorrea relacionada con la disfunción pancreática, y la otra mitad fueron evaluados por neutropenia 3. Posteriormente Agget et al 4 confirmaron los hechos previamente descritos y añadieron énfasis a las anomalías esqueléticas condrodisplásicas y anormalidades en las costillas.
Se trata de un trastorno multisistémico de herencia autosómica recesiva con una gran heterogeneidad en las manifestaciones clínicas. Presenta una incidencia de 1/100.000-1/200.000 nacidos vivos 5. Los rasgos centrales del síndrome son la disfunción pancreática exocrina y la disfunción de la médula ósea (sobre todo neutropenia). Otros rasgos que apoyan el diagnóstico son las anormalidades esqueléticas, hepatomegalia, elevación de transaminasas en suero, corta estatura e infecciones frecuentes.
Presentamos 2 casos clínicos cuyo motivo de remisión a nuestra consulta fue la hipertransaminasemia persistente.
Observación clínica
Caso 1
Niña de 2 años de edad remitida por su pediatra por hipertransaminasemia (a los 21 meses GOT: 108 y GPT: 181; a los 22 meses GOT: 133 y GPT: 212). Se trata de una RNP (34 semanas de gestación)/PEG III que había presentado adecuada curva pondoestatural hasta los 5 meses. Entre los 6 y los 7 meses presentó deposiciones malolientes sin productos patológicos. A los 10 meses inicia tratamiento empírico con enzimas pancreáticas que mantiene hasta los 2 años, aumentando progresivamente el peso. A los 18 meses presenta tres episodios de diarrea aguda, que se resolvieron con dieta exenta de proteína de vaca. Desde los 20 meses se instaura dieta libre, siendo las deposiciones de características normales aunque con olor agrio.
A la exploración física destaca un peso en percentil 3-10 y una talla en percentil 10, así como una hepatomegalia de 2 cm. Resto sin hallazgos patológicos.
Se le realiza un hemograma y bioquímica básica en la que se objetiva una fórmula leucocitaria con linfocitosis y neutropenia (730/μl), así como una GOT de 110 y una GPT de 124, siendo el resto de parámetros normales. Se descarta hepatopatía por virus hepatotropos más habituales, déficit de a1-antitripsina, enfermedad de Wilson, fibrosis quística, trastornos metabólicos más frecuentes, patología hepática autoinmune, miopatía oculta y enfermedad celíaca. Se solicita una ecografía abdominal (fig. 1) en la que se observa una hepatomegalia homogénea y un aumento de ecogenicidad en páncreas. Ante la sospecha de SDS se solicita una elastasa pancreática: 52 μg/g de heces (insuficiencia grave 6,7), una serie ósea que se informa como normal y una tomografía computarizada abdominal (fig. 2) donde se objetiva una disminución difusa y homogénea de la atenuación del páncreas, que muestra un tamaño en los límites bajos de la normalidad; los hallazgos se relacionan con una infiltración grasa global de la glándula.
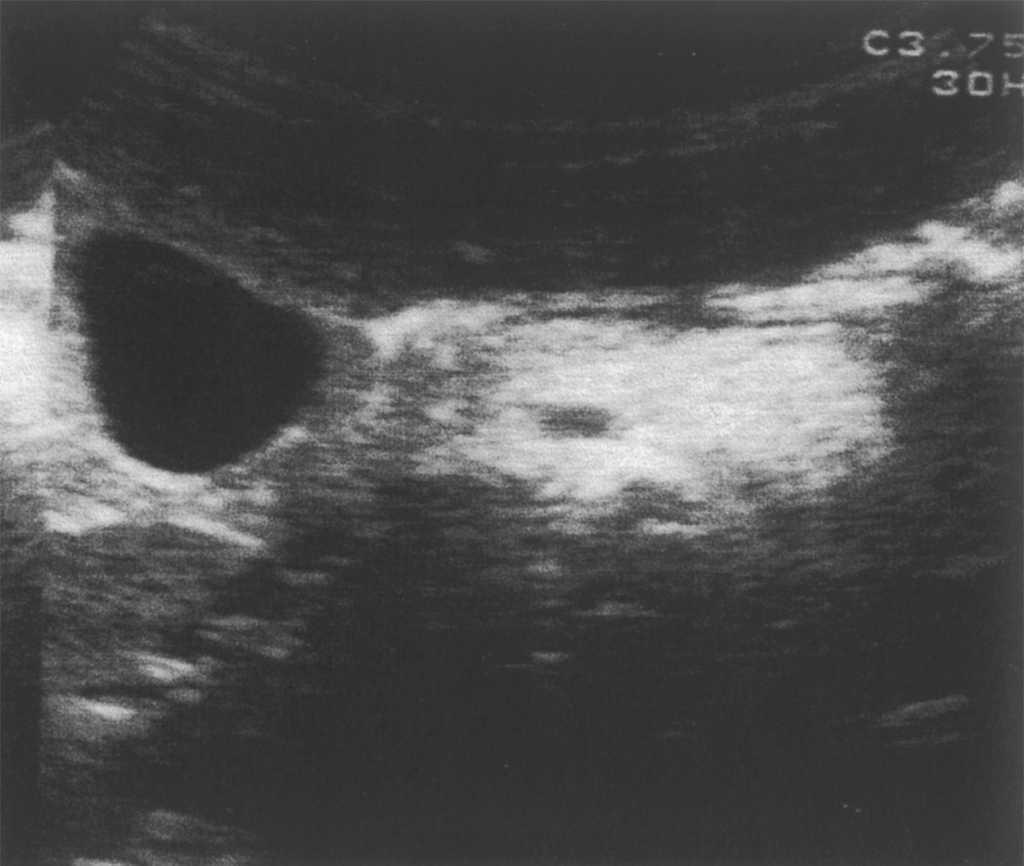
Figura 1. Aumento de ecogenicidad pancreática.
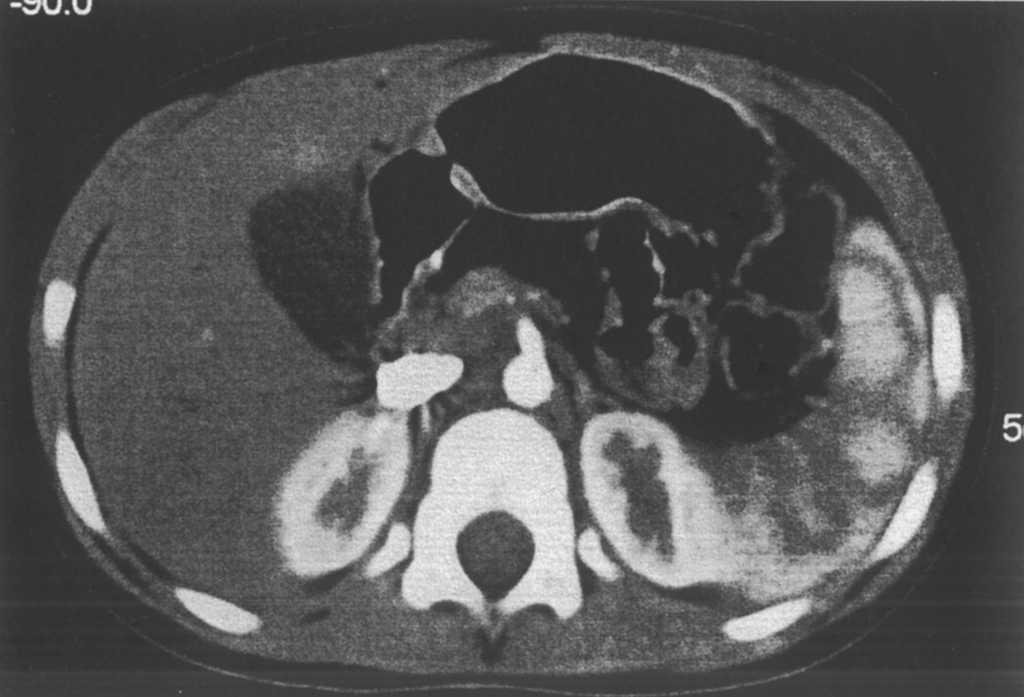
Figura 2. Disminución difusa y homogénea de la atenuación del páncreas, que muestran un tamaño en los límites bajos de la normalidad. Los hallazgos se relacionan con una infiltración grasa global de la glándula pancreática.
Durante su seguimiento, se ha detectado neutropenia en dos analíticas posteriores. Se ha resuelto la insuficiencia pancreática, alcanzando valores de elastasa normales a los 3 años y 9 meses. Las cifras de transaminasas han ido disminuyendo progresivamente, alcanzando valores normales a los 3 años y 4 meses. Así mismo, la curva pondoestatural se ha ido normalizando con velocidad de crecimiento normal.
Caso 2
Lactante de 17 meses remitida a nuestra consulta para biopsia hepática por hipertransaminasemia persistente detectada a los 3 meses y con GPT máxima (646) a los 12 meses. Entre los antecedentes destaca deposiciones malolientes y desligadas en número de 5 o 6 al día, así como una curva de peso en percentil 25 hasta los 6 meses, con escasa ganancia ponderal posterior.
A la exploración física destaca un peso y talla < P3 y un aspecto desnutrido.
Retrospectivamente en varios hemogramas había presentado neutropenia. Aportaba serología VHA, VHB, VEB, CMV y VIH negativa, así como test del sudor negativo (repetido en dos ocasiones). Se descartó hepatopatía autoinmune, déficit de a1-antitripsina y enfermedad de Wilson. Había presentado AAG (anticuerpos antigliadina) por encima de los valores normales en dos ocasiones por lo que se realizó una biopsia intestinal que fue normal. Había presentado un test de Van de Kamer patológico.
Ante la sospecha de SDS, se solicitó elastasa fecal: 1,4 μg/g (insuficiencia pancreática grave) y repetimos el test de Van de Kamer en el que nuevamente se apreció esteatorrea. Se solicitó ecografía abdominal en la que se apreció páncreas pequeño y ecogénico y una serie ósea que fue normal.
Durante su seguimiento presentó nuevos episodios de neutropenia así como valores de elastasa en rango de insuficiencia pancreática; las cifras de transaminasas fueron descendiendo progresivamente, alcanzando valores normales a los 2 años y 4 meses.
A los 2 años y 10 meses es remitida desde su hospital de origen con cuadro de púrpura fulminante coincidente con varicela. Ingresa con fallo multiorgánico y fallece a las 3 semanas.
Discusión
El SDS es un trastorno multisistémico autosómico recesivo, que fundamentalmente afecta la médula ósea, páncreas exocrino y placas de crecimientos de huesos largos. Se ha localizado recientemente su alteración en la región centromérica del cromosoma 7. El diagnóstico es problemático porque hay una considerable heterogenicidad clínica y ausencia de marcadores bioquímicos de la enfermedad. Variaciones en la gravedad de la enfermedad y manifestaciones clínicas complica el establecimiento de un diagnóstico definitivo 8.
Los criterios diagnósticos, tomados de Rothbaum y Perrault 9, se presentan en la tabla 1.

La hipertransaminasemia persistente puede ser una manifestación del SDS como en nuestros 2 casos. Es preciso descartar previamente tóxicos, virus hepatotropos, trastornos metabólicos, enfermedades de depósito (enfermedad de Wilson, déficit de α1-antitripsina), patología autoinmune, enfermedad celíaca, fibrosis quística y miopatía oculta.
La neutropenia cíclica (presente en el 80 % de pacientes con SDS) así como la insuficiencia pancreática se objetivó en ambos casos. Se desconoce la razón exacta del restablecimiento de la función pancreática con el tiempo. Se supone que estos pacientes incrementan la secreción de enzimas pancreáticas con la edad a causa del crecimiento del tejido pancreático residual 10.
Los pacientes más neutropénicos son los más propensos a sufrir infecciones graves. También se han visto defectos en la quimiotaxis. La trombocitopenia es la segunda anomalía más común, casi el 50 % de los pacientes con SDS muestran alteraciones en las tres líneas celulares. Este grupo es el más propenso a desarrollar leucemia mieloide aguda 11.
El aumento de ecogenicidad pancreática puede ser debido a microquistes en la fibrosis quística o a lipomatosis en el SDS 12.
Las alteraciones óseas (descritas en el 60 % de los pacientes con SDS) no han estado presentes en ninguna de nuestras pacientes, lo cual no excluye el diagnóstico. La disostosis metafisaria de los huesos largos (44 % de los pacientes) no suele manifestarse antes de los 6 años de edad. La distrofia torácica, que se caracteriza por engrosamiento costocondral, proyección de las costillas inferiores o una caja torácica estrecha, es menos común y se manifiesta antes de los 2 años de edad.
Durante la segunda mitad del primer año de vida, el ritmo de crecimiento disminuye, situándose el peso y la talla por debajo del P3. Este hallazgo lo encontramos en nuestras 2 pacientes. Con el tratamiento de la insuficiencia pancreática el ritmo de crecimiento mejora y muchos pacientes, aunque crecen a velocidad normal, nunca alcanzan un percentil normal. La estatura suele ser baja 13.
Correspondencia: Dra. Fátima Revert Lázaro.
Xavier Soler, 3, torre B, 9.º A. 03015 Alicante. España.
Correo electrónico: epmonjardin@hotmail.com
Recibido en mayo de 2005.
Aceptado para su publicación en enero de 2006.
