




Introducción
El hemangioendotelioma kaposiforme (HEK), descrito por Zuerkeberg en 1993 es un tumor solitario, de escasa incidencia y sin predilección por un sexo que nunca desaparece completamente ni provoca metástasis a distancia. Generalmente es grande, de forma excepcional aparece en vísceras o en localización craneofacial y cuando es mayor de 10 cm suele acompañarse de trombocitopenia. Presenta hasta el 50 % de mortalidad en la localización retroperitoneal.
El diagnóstico definitivo se establece por biopsia con el característico aspecto de zona de fibrosis con vasos dilatados y nódulo de células fusiformes que recuerdan a las del sarcoma de Kaposi pero sin infiltrado de células plasmáticas 1-16.
Observación clínica
Presentamos nuestra experiencia en el tratamiento de 4 casos de HEK de afectación torácica (una de sus localizaciones preferentes) que cursaron con síndrome de Kasabach-Merritt.
Caso 1
Paciente de pocas horas de vida con gran tumoración vascular que afectaba a tórax y abdomen (fig. 1) y que desarrolló una grave coagulopatía (4.000 plaquetas/μl) con hemotórax y hemorragia digestiva así como insuficiencia cardíaca congestiva que no respondían a tratamiento farmacológico y que provocaron el fallecimiento del recién nacido a las 8 h de vida. La necropsia puso de manifiesto alteraciones histológicas compatibles con hemangioendotelioma kaposiforme con afectación de tórax y abdomen.
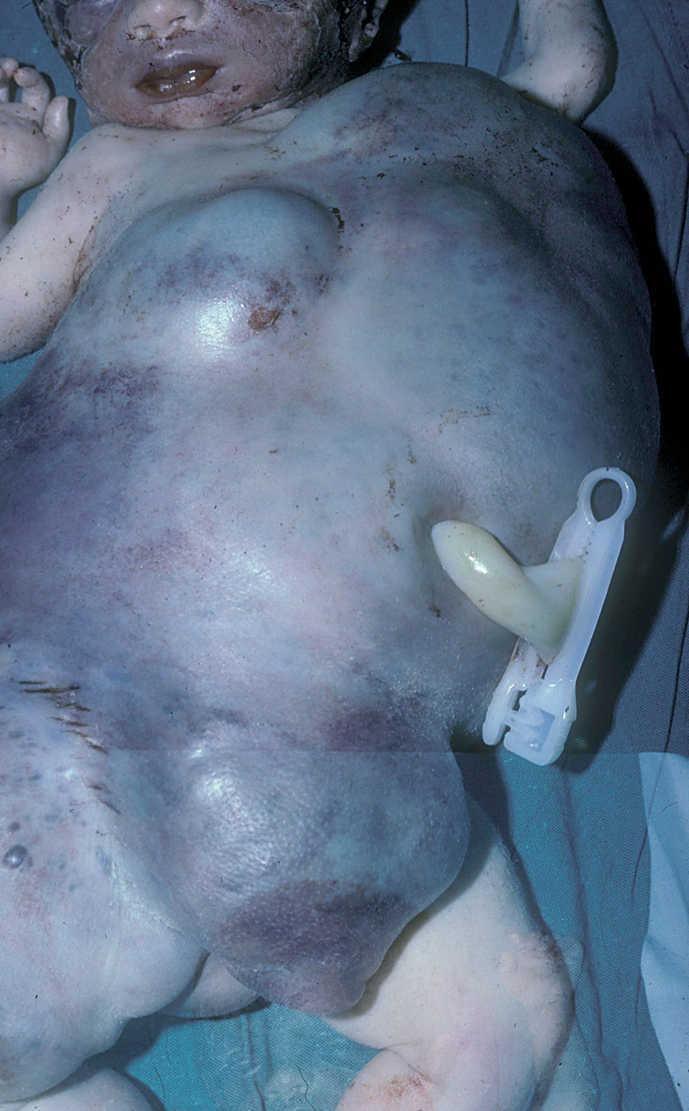
Figura 1.Caso 1.
Caso 2
Recién nacido con insuficiencia respiratoria grave, hemotórax que precisó drenaje pleural y gran tumoración en la parte superior del hemitórax izquierdo (fig. 2). La analítica muestra coagulopatía compatible con síndrome de Kasabach-Merritt (trombocitopenia intensa de 7.000 plaquetas/μl y elevación del dímero-D hasta 2 ng/ml). Se inició tratamiento con corticoides a 3 mg/kg/día sin obtener respuesta a los 5 días por lo que se inició la administración de interferón α2a a 3 millones de U/m 2 subcutáneo, siendo entonces la respuesta inmediata con una elevación progresiva de la cifra de plaquetas y una mejoría significativa en los parámetros ventilatorios con desaparición del hemotórax.
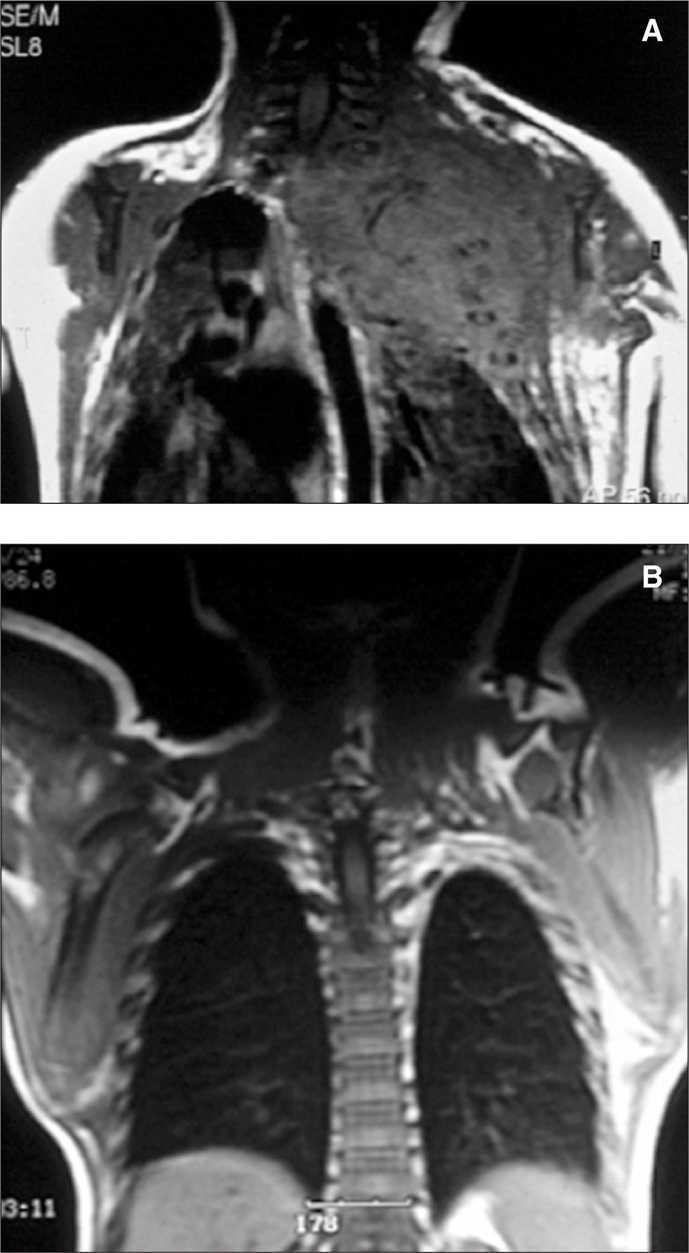
Figura 2.Caso 2.
A los 9 meses la RM mostraba una desaparición casi completa de la tumoración (fig. 2) y 8 años después el paciente sólo presentaba un leve síndrome de Claude- Bernard-Horner residual.
Caso 3
Paciente de 6 meses de edad con tumoración en hemitórax derecho (fig. 3) diagnosticada de hemangioma y que presentaba trombocitopenia que no respondía al tratamiento farmacológico con corticoides e interferón, por lo que se decidió la extirpación quirúrgica. En un primer tiempo, se llevó a cabo una resección incompleta de la tumoración que no consiguió corregir la coagulopatía, por lo que fue preciso un segundo abordaje quirúrgico que logró extirparla totalmente (fig. 3). A partir de este momento los parámetros analíticos iniciaron una elevación progresiva de la cifra de plaquetas hasta la normalización de esta. El diagnóstico histológico fue HEK.
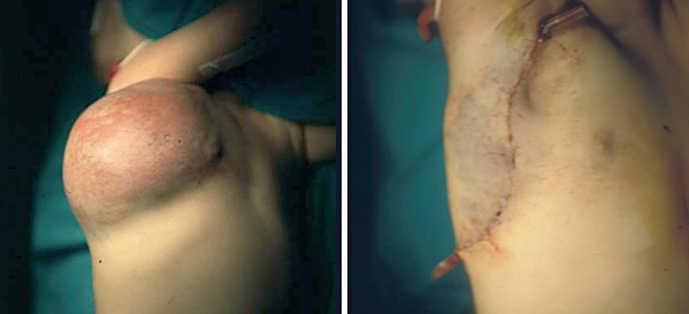
Figura 3.Caso 3.
Caso 4
Lactante de 4 meses con anomalía vascular cutánea en hemitórax y extremidad superior derecha diagnosticada erróneamente de "angioma plano" y que fue sometido a una sesión de láser de colorante pulsado. A las pocas horas, la niña fue empeorando, con insuficiencia respiratoria grave y hemotórax progresivo que precisaron intubación, tras la cual fue trasladada a la UCIP de nuestro centro. A su ingreso se practicó drenaje del hemotórax y se extrajo una analítica urgente que mostraba 6.000 plaquetas/μl y dímero D muy elevado (4 ng/ml). Se instauró tratamiento con corticoides, 3 mg/kg/día, sin obtener respuesta por lo que se sustituyen a las 3 semanas por interferón α2a a 3 millones/m 2 por vía subcutánea logrando una mejoría limitada a las 2 semanas de 26.000 y de 40.000 plaquetas/μl, 3 semanas más tarde. La paciente puede ser extubada y el hemotórax controlado. Ante la persistencia de la coagulopatía se inició tratamiento con vincristina, a 1 mg/m 2 subcutáneo semanal sin obtener respuesta. La paciente fue dada de alta hospitalaria en buen estado pero con trombocitopenia mantenida (fig. 4). Tres años después se inició terapia antiagregante con ácido acetilsalicílico y ticlopidina a 10 mg/kg/día ambas y administradas simultáneamente con lo que se consigue una elevación continuada y progresiva de la cifra de plaquetas hasta 210.000/μl y una reducción significativa del tamaño de la tumoración (fig. 4). Se realizó una biopsia de la tumoración que confirmó la sospecha clínica de HEK.
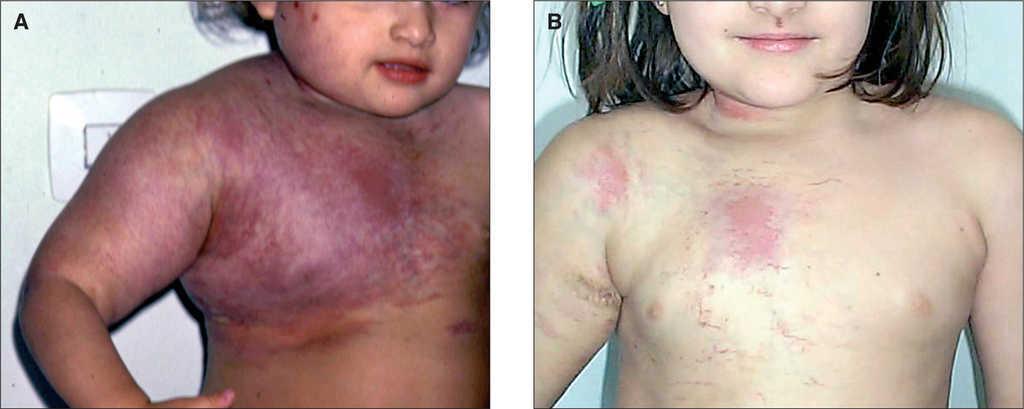
Figura 4.Caso 4.
Discusión
En la actualidad y al igual que en los casos presentados, casi todos los HEK son diagnosticados retrospectivamente, ante la aparición de un paciente con síndrome de Kasabach-Merritt. Aunque la evolución clínica de estos 4 niños ha sido diferente, todos han mostrado en algún momento alguno de los hallazgos característicos de este tumor vascular. Resulta imprescindible en estos casos establecer el diagnóstico diferencial correcto con la coagulopatía de consumo por estasis venosa que aparece en las malformaciones venosas (erróneamente diagnosticadas como "angiomas cavernosos"). En nuestros pacientes con HEK y síndrome de Kasabach-Merritt, la cifra de plaquetas fue sistemáticamente menor de 25.000/μl. Estos valores no se ven en la coagulopatía de las malformaciones venosas en las que las plaquetas superan la cifra de 75.000/μl 2,9,11,15.
Además, el síndrome de Kasabach-Merritt suele ser de aparición más precoz y frecuentemente neonatal (50 %).
En ocasiones el síndrome de Kasabach-Merritt es provocado por otro tumor vascular distinto del HEK, el angioma en penachos o angioblastoma de Nakagawa, pero cada vez son más las evidencias de que ambos tumores pertenecen a un mismo espectro histopatológico de origen similar y en diferente estadio evolutivo caracterizado en ambos casos por una rica proliferación linfática puesta de manifiesto por el anticuerpo monoclonal D2-40 en el estudio inmunohistoquímico 6-11.
Ninguno de los angioblastomas revisados en nuestro centro presentó trombocitopenia neonatal pero uno la desarrolló después de una infección respiratoria grave.
Sin embargo, y en presencia de coagulopatía, la biopsia puede ser de alto riesgo. En estas circunstancias, el aspecto violáceo con márgenes equimóticos, el curso clínico característico y los hallazgos radiológicos en la RM (tumor sólido de captación heterogénea con vasos dilatados con alto flujo a su través y que afecta simultáneamente a piel, tejido celular subcutáneo y músculo) son suficientes para establecer el diagnóstico e iniciar el tratamiento farmacológico.
Desde el punto de vista terapéutico, el tratamiento de elección y el único curativo es la extirpación radical de la lesión, pero esto sólo es posible en ocasiones sin provocar mutilaciones o secuelas funcionales y estéticas graves. El tratamiento farmacológico está basado en la tríada característica de agentes antiangiogénicos conocidos 12-16.
1.Corticoides. En dosis habituales de 2 mg/kg/día la tasa de respuesta es poco consistente (10 %). Por este motivo es habitual incrementar la dosis hasta los 5 e incluso los 10 mg/kg/día en caso de trombocitopenia grave y antes de considerar que no hay respuesta e iniciar el tratamiento con interferón.
2.Interferón α2a-b. La tasa de respuesta es del 50 al 60 % y se puede predecir detectando el factor básico de crecimiento de fibroblastos en orina (Uβ -FGF) previamente. A mayor elevación, mejor respuesta al interferón. No hay estudios que certifiquen mejor respuesta del interferón α2a o α2b. El riesgo de diplejía espástica debe ser considerado en tratamientos prolongados (de más de 6 meses) y la evaluación neurológica es por tanto imprescindible durante el tratamiento.
3.Vincristina (0,1 mg/kg/día). Dada la escasa respuesta a los corticoides en los tumores vasculares más graves, está siendo cada vez más usada como fármaco de primera o segunda elección.
4.Ciclosfosfamida (10 mg/kg/día). Representa otra alternativa de uso creciente en tumores vasculares de baja respuesta antiangiogénica.
5.Ticlopidina y ácido acetilsalicílico (10 mg/kg/día). Se han mostrado extremadamente eficaces en el control de la trombocitopenia que no responde a los anteriores fármacos.
En nuestros pacientes la toma de decisiones terapéuticas fue la correspondiente a las sospechas diagnósticas observadas mediante RM (que es la prueba de imagen más específica) y a la respuesta clínica en cada caso distinta. El único tumor resecable fue extirpado y no hubo efectividad de la terapia esteroidea ni con vincristina, siendo sólo uno de los tumores discretamente sensible al interferón.
La terapia intravascular sólo es útil como coadyuvante preoperatorio y no se utilizó. En los pacientes con tumores irresecables la embolización desarrolla arterias colaterales sin conseguir a largo plazo mejora alguna en los síntomas.
A pesar de la evidente respuesta de uno de los pacientes a la terapia antiagregante las mismas dosis no han sido eficaces en otros 2 pacientes con la misma enfermedad en localizaciones diferentes (brazo y pierna) tratados recientemente en nuestro servicio.
A pesar del progresivo conocimiento que tenemos de los distintos tumores vasculares, todavía estamos lejos de comprender algunos puntos clave de su origen, evolución clínica y respuesta diferente al tratamiento.
¿Qué factores y bajo qué condiciones favorecen la apoptosis celular programada? ¿Por qué esta involución es de meses en unos casos y de años en otros? ¿Dónde reside el mecanismo de respuesta o insensibilidad a diferentes agentes farmacológicos?
Es evidente que los avances del conocimiento molecular de los mecanismos íntimos de la angiogénesis ofrecerán respuesta a estas preguntas en el futuro 17,18.
La diferente evolución de cada uno de nuestros casos es fiel reflejo de la realidad clínica de este agresivo tumor.
En cualquier caso el éxito en el diagnóstico y tratamiento de esta entidad debe ir ligado al trabajo multidisciplinar de pediatras, patólogos, radiólogos y cirujanos, entre otros.
