





Introducción
El síndrome de Beckwith-Wiedemann (SBW) es una enfermedad genética que cursa principalmente con sobrecrecimiento físico durante los primeros años de vida. Los niños con SBW tienen elevado peso al nacimiento y crecimiento acelerado durante la primera infancia.
Estos pacientes suelen tener un desarrollo físico e intelectual dentro del rango de normalidad. Sin embargo, un pequeño porcentaje puede mostrar algún retraso en la maduración neurológica, provocado por alteraciones de la glucemia durante el período perinatal y/o anomalías cromosómicas de la región 11p (principalmente duplicaciones de origen paterno). Otro de los factores que hay que tener en cuenta es una susceptibilidad mayor que la población general a desarrollar cáncer.
El SBW se debe a alteraciones genéticas complejas (mecanismos epigenéticos que alteran el imprinting, pequeñas deleciones y duplicaciones, mutaciones concretas en genes de la región 11p, disomía uniparental, translocación y reordenamientos cromosómicos). La frecuencia de cada mecanismo patogénico es diferente y el diagnóstico molecular final se realiza, en general, en centros especializados en estas patologías.
El objetivo de este trabajo es presentar una guía clínica preventiva y anticipatoria para el seguimiento de pacientes con SBW, que contribuya a mejorar la calidad asistencial y la salud de esta población.
Características clínicas del SBW
Existe un número importante de hallazgos en el SBW. Los más frecuentes se enumeran en la tabla 1. Aunque no hay consenso absoluto sobre los criterios clínicos diagnósticos para el SBW, varios autores han sugerido en publicaciones diferentes criterios mayores y menores (tabla 2).


Características genéticas y subvariantes moleculares del SBW
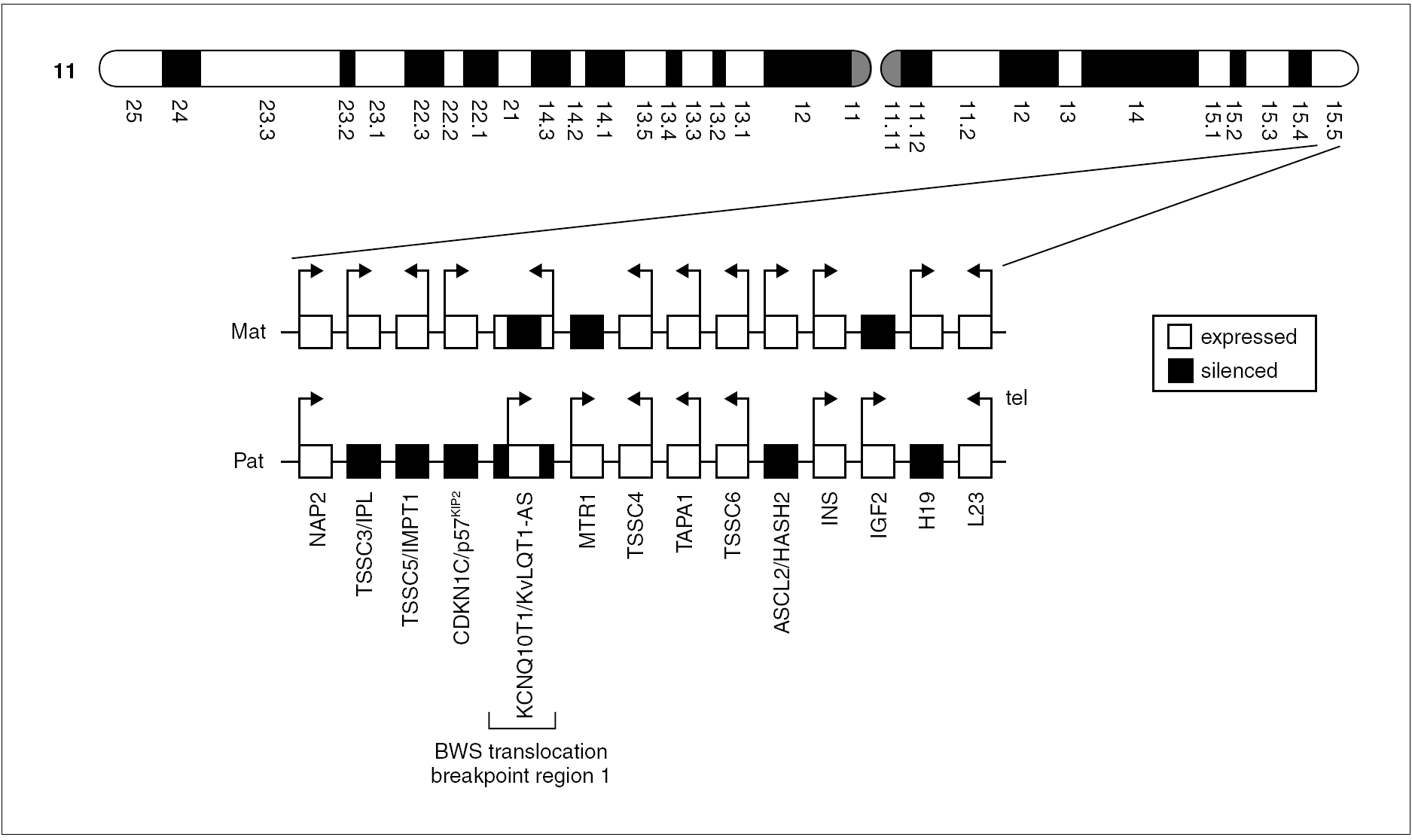
La tabla 3 presenta los distintos subgrupos genéticos y epigenéticos identificados en el SBW agrupados por su frecuencia relativa y muestra que es una entidad genéticamente heterogénea y con un efecto dependiente del origen parental de los genes heredados 1-6. En la figura 1 se observan algunos genes de la región 11p, activos o reprimidos en condiciones fisiológicas. A día de hoy hay 12 grupos o formas genéticas/epigenéticas diferentes, con distintas formas de herencia, diferentes genes y varios mecanismos moleculares implicados. La mayoría de los pacientes con SBW (alrededor del 90 %) son esporádicos y sin ninguna anomalía cromosómica demostrable (grupos A-E y H-J). El resto (10 %) muestran un patrón de herencia autosómico dominante con ligamiento en la región 11p15 (grupos F y G). Los reordenamientos cromosómicos relacionados con el SBW se observan en el 1-3 % de los casos (grupos I y J). Translocaciones e inversiones de la región 11p15 son típicamente de herencia materna (grupo J) mientras que las duplicaciones son de origen paterno (grupo I).

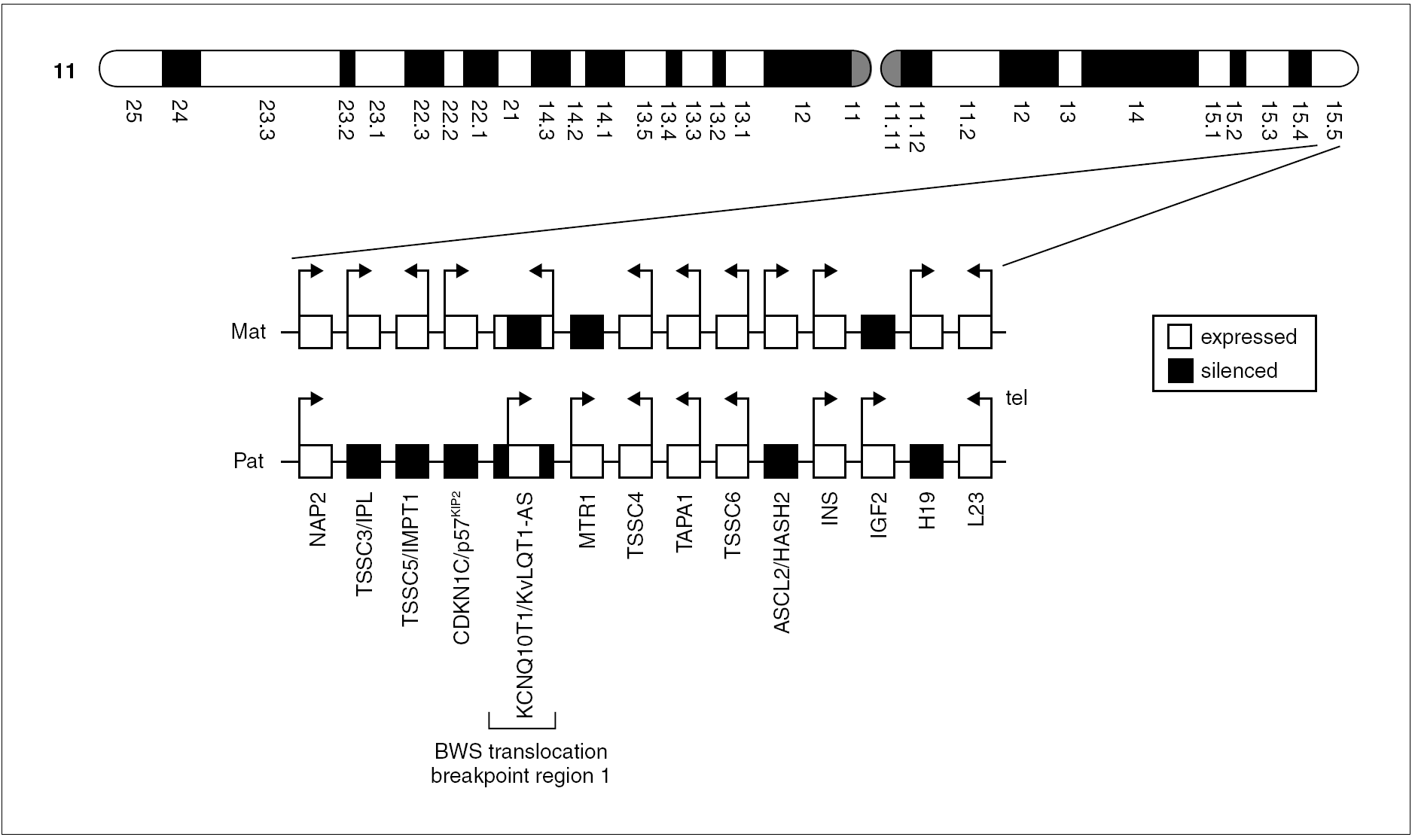
Figura 1. Orientación, expresión y ubicación de algunos genes de la región 11p15 implicados en la fisiopatología del SBW.
Las alteraciones genéticas y epigenéticas del SBW pueden afectar tanto a los genes supresores del crecimiento de origen materno como a los genes promotores del crecimiento de origen paterno. Aproximadamente el 10-20 % de los pacientes con SBW presentan disomía uniparental (DUP) paterna (grupo C). Todos los pacientes con DUP presentan mosaicismo somático, lo que demuestra que son necesarias ambas contribuciones parentales en la embriogénesis temprana y que la DUP aparece poscigóticamente, sólo se halla en algunos tejidos. En la DUP 11p15 paterna, el fenotipo de SBW y la predisposición tumoral es probablemente causada por la combinación de un aumento en la expresión de genes promotores del crecimiento (IGF2, de origen paterno) y una expresión disminuida o nula de genes supresores del crecimiento (como H19, de origen materno). Por último, el recientemente agregado grupo L incluye aquellos pacientes con microdeleciones del gen KCNQ1OT1 (LIT1) o del centro de imprinting de los genes IGF2/H19, cuya frecuencia exacta es aún desconocida.
CUIDADOS MÉDICOS GENERALES RECOMENDADOS PARA LOS PACIENTES CON SBW
Evaluación prenatal
Evidencias
Los embarazos múltiples tienen un riesgo sensiblemente mayor de aparición de SBW 7. Este se ve notablemente incrementado en los embarazos obtenidos con técnicas de reproducción asistida, en especial por fecundación in vitro o inyección intracitoplasmática. Según datos disponibles en la literatura médica, las técnicas de reproducción asistida incrementan entre 4 y 5 veces el riesgo de aparición de SBW respecto a la población general 8-14. Por tanto, si la frecuencia del SBW en la población general es de aproximadamente 1:14.000, entre los nacidos gracias a técnicas de reproducción asistida estaría en torno a 1:3.000.
Los fetos portadores de SBW pueden tener hallazgos ecográficos prenatales sugestivos de la enfermedad. Hallazgos habituales son polihidramnios, onfalocele y macrosomía, cuya frecuencia justifica un seguimiento estricto del embarazo y la programación del momento del parto, que en la mayoría de los casos acaba en cesárea programada 15-26.
Conducta
Si hay sospecha prenatal de SBW:
1.Considerar seguimiento por un equipo obstétrico experimentado y, si fuera posible, familiarizado con el SBW.
2.Considerar la realización de ecografías dismorfológicas, evaluando con detalle la anatomía fetal, en particular la región craneofacial y el abdomen, en el que se medirán los órganos sólidos.
3.Programar una cesárea si existe onfalocele o macrosomía fetal importante.
4.Alertar al equipo neonatal del posible diagnóstico, para que se tomen medidas que eviten la hipoglucemia neonatal.
Evaluación clínica posnatal
El neonatólogo o pediatra debe estar pendiente de los posibles hallazgos y/o complicaciones de los recién nacidos con SBW. Como dichos hallazgos pueden ser diversos, de gravedad variable, y diferentes según la edad de los pacientes, hemos dividido el seguimiento clínico por edades.
Nacimiento y primer año de vida
Evidencias
Los neonatos con SBW presentan riesgo de hipoglucemia, por lo que la concentración de glucosa en sangre se debe evaluar de forma seriada para prevenir este importante problema 27,28.
La mayoría de los niños no presentan retraso en la maduración neurológica, excepto un pequeño grupo de pacientes que han sufrido hipoglucemias insuficientemente tratadas o aquellos que presentan una anomalía citogenética (duplicación de origen paterno) en la región 11p.
El riesgo de tumores está incrementado cuatro veces en pacientes con hemihipertrofia o nefromegalia, y aparecen en el abdomen en el 95 % de los casos y antes de los 4 años de vida. La mayoría de los tumores asociados al SBW son embrionarios y eventualmente susceptibles de ser diagnosticados con marcadores bioquímicos y/o mediante ecografía. El cribado de tumores en los primeros años de vida permite un diagnóstico precoz de cáncer, lo cual mejora la supervivencia de estos pacientes a corto y largo plazo 29-37 (tablas 4 y 5).


La macroglosia sintomática que cursa con dificultad respiratoria, infecciones incontrolables de la vía área superior, dificultad para alimentarse, etc., mejora con el tratamiento quirúrgico de reducción 38-48.
Conducta
Examen físico
Una vez sospechado y/o confirmado el diagnóstico de SBW, se deben realizar las siguientes acciones:
1.Hacer examen físico completo incluyendo exploración abdominal completa en la primera consulta.
2.Examinar y supervisar el proceso de duelo y adaptación de los padres al diagnóstico de su hijo, evaluando la necesidad de apoyo psicológico profesional a los padres.
3.Continuar los exámenes clínicos detallados incluyendo palpación abdominal cuidadosa cada 3-4 meses.
4.Consignar peso, talla y perímetro cefálico en cada consulta, y realizar una curva con cada uno de estos parámetros.
5.Evaluar la alimentación tanto en cantidad como en calidad.
6.Comprobar si existe macroglosia, la efectividad de la succión y de la deglución y el tratamiento de las secreciones y de la vía aérea.
7.Evaluar la presencia y/o aparición de hernias umbilicales o inguinales.
8.Realizar exploración cardiológica. Si se constata miocardiopatía hipertrófica neonatal, continuar el seguimiento con el cardiólogo infantil, incluyendo radiografía de tórax anual.
9.Evaluar la función neurológica y el desarrollo psicomotor durante el primer año de vida.
Estudios complementarios
1. Realizar seguimiento riguroso y seriado de la glucemia en el período neonatal.
2. En caso de hipoglucemia persistente, descartar hiperinsulinismo mediante aproximación diagnóstica por etapas. Considerar consulta al endocrinólogo infantil. Descartar nesidioblastosis y/o implantación de medicación.
3. Realizar cariotipo, estudios de hibridación in situ con fluorescencia (FISH) para la región 11p y estudios para descartar DUP. Si estos son normales, hay que contactar con un centro especializado para realizar el diagnóstico molecular (aunque estos resultados pueden demorarse, ya que muchos de dichos centros los realizan como estudios de investigación).
4. Realizar trimestralmente determinación de alfafetoproteína, gonadotropina coriónica y catecolaminas en sangre.
5. Realizar ecografías abdominales trimestrales para el diagnóstico precoz de tumoración abdominal oculta.
6. Realizar estudio de orina completa cada 3 meses para detección precoz de tumor de Wilms.
7. Realizar radiografía de tórax semestralmente.
8. Realizar ecografía cerebral antes del alta neonatal mientras la fontanela esté permeable.
9. Realizar tomografía computarizada (TC) o resonancia magnética (RM) de abdomen si existe nefromegalia o imagen sospechosa abdominal o torácica.
10. Evaluar la dentición con consulta al odontólogo.
Otros aspectos
1.Solicitar consulta de asesoramiento genético con un genetista. Eventualmente se completarían los estudios con análisis moleculares del niño y sus padres.
2.Planear soporte educacional y psicológico de la familia.
3.Si presenta retraso psicomotor comenzar con estimulación precoz.
4.Si existe macroglosia sintomática (dificultad ventilatoria, dificultad en la alimentación o infecciones respiratorias altas a repetición), consultar con el especialista la posibilidad de realizar glosectomía parcial.
Entre 1 y 4 años de vida
Evidencias
El cribado de tumores en los primeros años de vida permite un diagnóstico temprano de cáncer y mejora la supervivencia a corto y largo plazo de estos pacientes.
El riesgo de tumores está incrementado cuatro veces en aquellos pacientes con hemihipertrofia o nefromegalia.
La edad media de la mayoría de los niños con SBW que desarrollarán un tumor está alrededor de los 2 años 29-37.
Conducta
Examen físico
1.Realizar examen físico completo incluyendo evaluación abdominal exhaustiva en cada consulta.
2.Evaluación seriada del desarrollo psicomotor. Puede haber problemas en el área motórica gruesa (inicio de la marcha, saltar, girar, correr, galopar). Considerar evaluación por el neurólogo infantil si existe hipotonía o si hay alteraciones persistentes de desarrollo psicomotor.
3.Continuar los exámenes clínicos detallados incluyendo palpación abdominal rigurosa cada 3-4 meses.
4.Consignar peso, talla y perímetro cefálico en cada consulta y completar cada una de las curvas de crecimiento somatométrico.
5.Puede haber erupción precoz de la dentición y debe informarse a la familia de que esto es esperable por ser un hallazgo frecuente en el SBW.
6.Es imprescindible contar con un estudio de neuroimagen (TC o RM cerebral) en forma periódica (al menos cada 2 años).
7.Evaluar la alimentación tanto en cantidad como en calidad.
Estudios complementarios
1.Realizar cariotipo, estudios de FISH para la región 11p y estudios moleculares que incluyan el estudio de DUP si no se hubieran realizado antes.
2.Realizar trimestralmente determinación sérica de alfafetoproteína, gonadotropina coriónica y catecolaminas urinarias.
3.Realizar ecografías abdominales cada 3-4 meses para diagnóstico precoz de tumoración abdominal oculta.
4.Realizar estudio de orina completa cada 3-4 meses.
5.Realizar control de la edad ósea anualmente.
6.Realizar radiografía de tórax cada 6 meses.
7.Realizar al menos otra ecografía cerebral mientras la fontanela esté permeable.
8.Realizar TC completa si existe nefromegalia o imagen sospechosa abdominal o torácica.
9.Evaluar la dentición con consulta al odontólogo.
10. Si la evolución del lenguaje no es la esperada, realizar una audiometría o potenciales evocados auditivos.
Otros aspectos
1.Solicitar, si es factible, consulta de asesoramiento genético con un genetista. Si aún no se han realizado, se completará el estudio con análisis moleculares del niño y sus padres.
2.Evaluar la necesidad de ofrecer soporte educacional y psicológico a la familia.
3.Si presenta retraso psicomotor comenzar con estimulación precoz.
4.Considerar consulta con el endocrinólogo infantil.
Entre los 4 y 10 años de vida
Evidencias
El riesgo de desarrollar tumores en los niños con SBW mayores de 4 años disminuye considerablemente con relación a edades más precoces. Casi el 95 % de los pacientes con SBW y tumores son menores de 4 años. Esta información debe ser transmitida a la familia para disminuir la ansiedad que genera la posibilidad de que su hijo sea incluido en un programa de seguimiento para detección de tumores 29-37.
Conducta
Examen físico
1.Realizar examen físico completo incluyendo evaluación abdominal completa en cada evaluación anual.
2.Continuar los exámenes clínicos detallados incluyendo palpación abdominal rigurosa anualmente.
3.Consignar peso, talla y perímetro cefálico en cada consulta. Realizar una curva con cada uno de estos parámetros.
4.Evaluar la alimentación tanto en cantidad como en calidad.
5.Considerar una evaluación psicopedagógica previa al ingreso en educación primaria si persiste el retraso de la maduración psicomotora.
Estudios complementarios
1.Realizar cuantificaciones anuales de la glucemia.
2.Realizar semestralmente determinación sérica de alfafetoproteína, gonadotropina coriónica y catecolaminas.
3.Realizar ecografías abdominales de forma anual para diagnóstico precoz de tumoración abdominal oculta.
4.Realizar estudio de orina completa anualmente.
5.Realizar radiografía de tórax una vez al año.
6.Realizar TC abdominal si existe nefromegalia o imagen sospechosa en la región abdominal o torácica.
7.Evaluar la dentición con consulta al odontólogo.
Otros aspectos
1.Solicitar, si es factible, consulta de asesoramiento genético con un genetista.
2.Eventualmente se completarían los estudios con análisis moleculares en el niño y sus padres.
3.Soporte educacional y psicológico a la familia.
Entre los 10 años de vida y la primera etapa de la juventud
Evidencias
Aunque existen casos aislados de aparición de tumores en pacientes con SBW durante la segunda década de la vida, el riesgo disminuye de forma considerable a partir de esta edad, y es innecesario un programa específico de seguimiento.
Conducta
Examen físico
1.Realizar examen físico completo incluyendo una exploración abdominal cuidadosa en cada visita anual.
2.Consignar peso, talla y perímetro cefálico en cada consulta. Realizar una curva en cada uno de estos parámetros.
3.Evaluar la alimentación tanto en cantidad como en calidad.
4.Considerar apoyo psicológico del paciente. Los niños y adolescentes con SBW tienen una autoestima baja, les cuesta relacionarse, pueden tener cambios incontrolados de carácter y son más vulnerables a daño emocional por parte de sus compañeros.
5.Considerar apoyo escolar continuo.
6.Evaluar su conducta para descartar posibles alteraciones psiquiátricas o neurosis.
7.Alertar a los padres sobre la posibilidad de traumatismos en la práctica de deportes debido a la torpeza motora.
Estudios complementarios
1.Realizar anualmente hemogramas (descartar enfermedades linfoproliferativas) y ecografías abdominales (tumoraciones abdominales).
2.Realizar estudio de orina completa anualmente.
3.Evaluar la columna vertebral para descartar escoliosis y cifosis.
4.Realizar radiografía de tórax cada año.
5.Evaluar la dentición con consulta al odontólogo.
6.Considerar realizar un estudio de neuroimagen (TC o RM) cada 2-3 años.
Otros aspectos
1.Si no se hubiesen realizado, completar el estudio con análisis moleculares en el niño y sus padres.
2.Ofrecer soporte educacional y psicológico a la familia.
Adultos con SBW
Conducta
Examen físico
Realizar examen físico completo anual.
Estudios complementarios
Planear la realización anual de un estudio completo de laboratorio, incluyendo enzimas y marcadores tumorales.
Otros aspectos
Insistir en los mecanismos de transmisión de la enfermedad y el riesgo en futuros hijos del paciente, si se ha podido determinar la subvariante molecular del SBW.
Nota aclaratoria
Esta guía pretende ser orientativa y en ningún caso estricta o única. Si bien ha sido elaborada en forma rigurosa, puede y debe ser adaptada a cada paciente en particular. La medicina es una ciencia cambiante y evolutiva y las recomendaciones que aquí ofrecemos pueden y deben ser actualizadas periódicamente para asegurar un correcto seguimiento en salud de los pacientes con síndrome de Beckwith-Wiedemann.
Este documento ha sido elaborado en el seno de la Comisión de Genética Clínica de la Red de Centros de Genética Clínica y Molecular (C03/07) del Fondo de Investigación Sanitaria, Instituto de Salud Carlos III, Ministerio de Sanidad y Consumo.
Correspondencia: Dr. P. Lapunzina Badía.
Servicio de Genética Médica. Hospital Universitario La Paz.
P.º de la Castellana, 261. 28046. Madrid. España.
Correo electrónico: plapunzina.hulp@salud.madrid.org
Recibido en octubre de 2005.
Aceptado para su publicación en diciembre de 2005.
