



La hiperamonemia sintomática en el recién nacido es una urgencia médica que debe reconocerse de manera precoz, diagnosticarse de manera específica y tratarse de forma intensiva para mejorar el pronóstico inmediato y a largo plazo de estos niños.
Para esto, es necesario que el pediatra en general y el neonatólogo en particular tengan presente una secuencia diagnosticoterapéutica de actitud inmediata que contribuya a una adecuada actuación.
Symptomatic hyperammonaemia in newborn is a medical emergency that should be recognised in its early stages, specifically diagnosed and aggressively treated to improve the immediate and long-term prognosis of these children. The paediatrician and the neonatal doctor should have a diagnosis-therapy scheme for its urgent management.
La hiperamonemia conlleva una encefalopatía rápida y grave, a la que el recién nacido (RN) es especialmente vulnerable. Por esto, se considera útil disponer de una guía práctica de diagnóstico y tratamiento inicial en el RN.
Definición de hiperamonemiaSe considera hiperamonemia cuando el RN presenta valores de amonio mayores de 110μmol/l (190μg/dl)1,2.
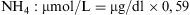
Ante elevaciones moderadas (entre 110 y 150μmol/l), debe repetirse la muestra si una enfermedad de base no la explica claramente. Si los valores de amonio son superiores a 150μmol/l, debe llevarse a cabo una evaluación diagnóstica específica para la hiperamonemia.
Si el RN se encuentra en un hospital comarcal y presenta valores de amonio de 150μmol/l o más, debe realizarse su traslado inmediato a un centro de referencia.
La toma de la muestra debe realizarse por vía venosa o arterial, sin manguito; se recomienda utilizar una vía de calibre grueso para que no haya hemólisis. La sangre debe depositarse en un tubo con EDTA (ethylene diamine tetra-acetic ‘ácido edético’) en vacío para que no tome contacto con el aire del ambiente, debe conservarse en hielo y analizarse inmediatamente, ya que en una hora el amonio sube y se sintetiza en células sanguíneas.
SintomatologíaRN normal que a las pocas horas o días presenta como primeros síntomas succión débil, hipotonía, vómitos, letargia progresiva, respiración irregular y, en algunos casos, convulsiones. Esta sintomatología progresa rápidamente al coma y a la muerte temprana. De hecho, el comienzo clásico de una hiperamonemia metabólica neonatal es un coma súbito y potencialmente mortal.
Evaluación diagnóstica3El objetivo de esta evaluación es llegar al diagnóstico correcto y administrar a tiempo el tratamiento apropiado para evitar la muerte o el daño permanente en el cerebro del RN.
- –
Los estudios iniciales están disponibles en los análisis de laboratorio habituales. Se trata de un perfil metabólico sencillo sobre la situación del sujeto, que pude además orientar a una valoración sobre qué estudios más específicos deben hacerse en primer lugar. Entre éstos se encuentran valores de amonio, equilibrio acidobásico, sodio, cloruro, transaminasas, CK (creatin kinase ‘creatincinasa’), análisis de coagulación y lactato. También puede evaluarse la presencia de cetonas en orina o cuerpos cetónicos (ß hidroxibutirato) en una muestra capilar (Optium Xceed®) y calcularse la brecha aniónica.
- –
Las investigaciones más específicas para el diagnóstico diferencial consisten tanto en la realización de las tomas antes de iniciar el tratamiento (si el sujeto no hiciera ninguna micción antes de iniciarlo, la orina podría tomarse durante las primeras horas del tratamiento) como en la preparación de las muestras y su congelamiento hasta que se remitan a laboratorios especializados.
Según el laboratorio, las tomas pueden hacerse de la siguiente manera:
- –
Para la valoración de los aminoácidos, las acilcarnitinas y la actividad de la biotinidasa, se colocan 3 cc de sangre en un tubo con gel, se centrifugan, se separa el suero y luego se congela.
- –
Para la valoración de los aminoácidos, se colocan 2 cc de sangre en un tubo con EDTA, se centrifugan, se separa el plasma y luego se congela. Para la valoración de las acilcarnitinas y la actividad de la biotinidasa, se coloca la sangre total en un papel de filtro S&S.
- –
Para la valoración de los aminoácidos, los ácidos orgánicos y especialmente el ácido orótico en orina, sirve la 1.a micción que se realiza.
- 1.
El déficit del ciclo de la urea4 es la causa más frecuente de las hiperamonemias graves, ya que representa el 60% en el período neonatal. Generalmente, se presenta con alcalosis respiratoria y cifra baja de urea. Sin embargo, puede aparecer con alcalosis o acidosis metabólica.
Tabla 1.Hiperamonemia neonatal. Diagnóstico diferencial
Acidosis metabólica Amonio (N<110μmol/l) Lactato (N<2,5mM, <20mg/dl) GOT–GPT (N<40U/l) CK (N<190U/l) Ácido úrico (N<7mg/dl) 3-0Hbutirato. Si es >0,4mM hay síntesis de cetónicos. En RN<0,4 es normal Si hay hipoglucemia (glucemia venosa<45mg/dl) 3-0H-3metil glutárico aciduria Grave N o A N o A A A N B (<0,3μmol/l) Trastornos de la betaoxidación de los ácidos grasos Moderada o grave N, A (> 300μmol/l) o muy A A A A N o A B Defecto complejo II de cadena respiratoria. (MADD) (GLUT II) Grave A A A A A B Hiperinsulinemia+hiperamonemia No <300μmol/l N N N N Siempre<0,3 Si no hay hipoglucemia Trastornos del ciclo de la urea No o leve Muy A N o A A N N N Jarabe de arce Leve o moderada N o levemente A N o levemente A N N N N o A Acidurias isovaléricas y 3 metil crotónica (catabolismo leucina) Moderada o grave 200-700 A N o A N N o A A Acidemias propiónica y metilmalónica Grave Muy A A N o A A N o A Muy A para RN (>1mM) Déficit multiple de carboxilasas Moderada o grave 90-700 A N o A N N o A A Atrofia girata Leve Muy A N N o A N N <0,3 HHH (pH orina >7) Leve 200-350 N N N N <0,3 Defectos de transporte de aminoácidos dibásicos (cistinuria: pH orina >7,5) Leve 120-250 N N N N <0,3 A: alto; B: bajo; CK: creatin kinase ‘creatincinasa’; GOT: glutamic oxaloacetic transaminase 'transaminasa glutamicoxalacética'; GPT: glutamic pyruvic transaminase 'transaminasa glutamicopirúvica'; HHH: hiperamonemia, hiperornitinemia, homocitrulinuria; N: normal; RN: recién nacido; vsp: volumen de sangre pulmonar.
- 2.
La acidurias orgánicas representan el 30% de las hiperamonemias neonatales graves. Habitualmente se presentan con acidosis metabólica con hiato aniónico elevado, hiperlactacidemia y cetosis o cetonuria. Se exceptúan las acidemias por alteración en la oxidación de ácidos grasos y en la 3-0H-3metil glutárico aciduria, en la que no hay síntesis de cetónicos.
- 3.
El déficit de la betaoxidación de los ácidos grasos, sobre todo los de cadena larga, puede presentarse con una hiperamonemia marcada. Generalmente, se presenta con hipoglucemia, hipocetosis o elevación de la CK.
- 4.
La atrofia girata es un trastorno del metabolismo de la ornitina que se concentra en la mitocondria y bloquea el transporte de la arginina y de la ornitina. Ésta puede condicionar hiperamonemias de hasta 1.800μmol/l.
- 5.
Los trastornos en el transporte de los aminoácidos dibásicos en las cistinurias pueden producir una pérdida de la L-Arginina, lo que puede conducir a una hiperamonemia.
- 6.
El síndrome de hiperamonemia, hiperornitinemia y homocitrulinuria es un defecto del transporte mitocondrial de la ornitina que origina una deficiencia intramitocondrial y, como consecuencia, un trastorno del ciclo de la urea.
- 1.
La hiperamonemia transitoria del RN se origina por la falta del cierre inmediato del conducto venoso de Arancio después del nacimiento al derivar el flujo que la sangre porta al hígado. Se presenta entre las primeras 24 a 36h de vida y es más frecuente en los RN con distrés respiratorio. En éstos el cociente de glutamina y de NH4 es menor de 1,6μmol/μmol. El aumento de los valores de amonio en este trastorno se resuelve generalmente entre 4 a 5 días, pero si los valores de amonio son mayores de 250μmol/l, los RN pueden precisar tratamiento.
- 2.
Las transaminasas elevadas o el tiempo de protrombina alargado de la insuficiencia hepática primaria grave, pueden observarse en otras causas metabólicas, aunque generalmente en menor intensidad. Asimismo, es frecuente que haya elevación de aminoácidos plasmáticos (fenilalanina, tirosina, aminoácidos de cadena ramificada).
- 3.
Otros trastornos pueden ser la asfixia perinatal o cualquier enfermedad grave. Generalmente estos valores no se presentan tan elevados.
Tratamiento médico (fig. 1 y tabla 2)
Ante una hiperamonemia documentada con valores de amonio superior a 180μmol/l y sin saber el diagnóstico en las primeras 2h debe procederse de la siguiente manera:
- –
Cese del aporte proteico y alto aporte calórico: las necesidades basales deben incrementarse al menos en un 25% para frenar el catabolismo y estimular el anabolismo, y así revertir la degradación de las proteínas endógenas. Esto se consigue con infusiones hiperosmolares de glucosa (10mg/kg/min de glucosa), asociando insulina si la glucemia es elevada. Se pueden utilizar emulsiones lipídicas siempre que no se sospeche un trastorno de la betaoxidación de los ácidos grasos. Se aconseja nutrición parenteral total en la fase inicial, mientras se precise un alto aporte energético de glucosa y de técnicas invasivas para la depuración exógena del tóxico. La nutrición enteral debe iniciarse tan pronto sea posible.
- –
La L-Arginina es indispensable ya que sin ésta el ciclo de la urea no funciona. Debe suministrarse una dosis de carga de 350mg/kg, por vía intravenosa en 90min, luego debe mantenerse una dosis de 600mg/kg/día en perfusión continua. La preparación requiere 1g en 50ml de glucosa al 10%.
No es habitual encontrar en el mercado un preparado para la administración por vía intravenosa. La alternativa es la administración por vía oral a 500mg/kg/día.
Una vez que se tenga el diagnóstico definitivo, si se trata de un déficit enzimático intramitocondrial del ciclo de la urea (carbamil fosfato sintetasa y ornitin carbamil transferasa), es suficiente una dosis de 150 a 200mg/kg/día. Las dosis altas de arginina pueden producir acidosis metabólica hiperclorémica.
- –
Con el fin de «lavar» el exceso de nitrógeno sanguíneo se utilizan quelantes del amonio, como benzoato, fenilacetato y fenilbutirato.
- –
El benzoato sódico puede prepararse en fórmula magistral. Se suministra una dosis de carga de 250mg/kg por vía intravenosa en 90 min, seguida por perfusión de 250 a 500mg/kg/día. La preparación requiere 1g en 50ml de glucosa al 5 o 10% en un frasco de vidrio protegido de la luz.
Debido a que no se puede emplear una mayor concentración, puede hacerse una dilución común con arginina y benzoato en 50ml de glucosa al 10% para evitar la administración de volúmenes altos de líquidos en el RN.
- –
La administración de la dosis del fenilbutirato (Ammonaps®, comprimidos de 500mg) debe ser de 200 a 600mg/kg/día por vía oral.
Se han descrito algunos efectos secundarios en el uso de estos quelantes del amonio, como mucositis, acidosis o alcalosis, hipoalbuminemia, hipopotasemia y conjugación deficiente de la glicina (en el caso del benzoato), lo que puede incrementar la aparición de naúseas y vómitos.
- –
En la actualidad se puede disponer de la administración de benzoato sódico más fenilacetato sódico intravenoso (Ammonul®, que es un medicamento extranjero de Swedish Orphan). Cada centímetro cúbico contiene 100mg de benzoato y 100mg de fenilacetato sódico, y debe diluirse previamente al 10% en suero glucosado. Se utiliza una dosis de carga de 250mg/kg de fenilacetato y de benzoato entre 90 a 120 min y se continúa con otra dosis iguales hasta las 24h de tratamiento.
- –
La carnitina (Carnicor® viales) se administra en una dosis de carga de 50mg/kg en 90 min. Posteriormente, se mantiene a 100mg/kg/día, repartido en varias dosis al día.
Ante la sospecha de acidemias orgánicas hay que valorar el uso de los quelantes del amonio, especialmente del benzoato, debido a un posible riesgo de depleción intramitocondrial de la coenzima A. Tampoco se debe emplear carnitina si se sospecha un trastorno de la betaoxidación de los ácidos grasos debido al el riesgo de aumentar la producción de acilcarnitinas tóxicas.

Cofactores empleados en el tratamiento agudo del recién nacido con hiperamonemia sin diagnóstico etiológico
| Cofactor | Modo de acción | Enfermedad o déficit enzimático en el que ayuda | Dosis | Observaciones |
| L-Arginina | En hiperamonemias de cualquier etiología, excepto en el déficit de arginasa. Es indispensable, ya que activa el ciclo de la urea (única vía para metabolizar el amonio) | Activador de la NAGS, (1.a enzima del ciclo de la urea para la sintesis de NAG). Activador de la 2.° enzima del ciclo de la urea | 200-700mg/kg/día por vía intravenosa o al 10% por sonda a débito continuo | Los déficits de NAGS no se activan con L-Arginina |
| NCG (Carbaglu®) | Análogo del NAG, activador natural de la 2.° enzima (CPS) del ciclo de la urea. La CPS trasforma el amonio (tóxico) en carbamil fosfato (no tóxico) y se activa tanto con NAG como con NCG | Se emplea en déficits de NAGS, en deficiencia de CPS sensible a NCG y en hiperamonemias por inhibición de NAGS: (acidemias orgánicas, déficits de L-Arginina, trastornos de la betaoxidación, hiperinsulinismo, hiperamonemia y otras) | 100-200mg/kg/día | Disminuye el amonio fuertemente en déficits de NAGS. Es muy efectiva en sujetos con NAGS inhibida |
| Biotina (Medebiotin forte®) | Cofactor de carboxilasas | Déficit múltiple de carboxilasas y de biotinidasa | 30mg/día | Muy efectiva |
| Hidroxicobalamina (Megamilbedoce®) | Precursor de los cofactores de la metilmalonil CoA mutasa y de la remetilación de la homocisteína | MMA + homocistinuria | Intramuscular: 1mg/día. Vía oral: 10mg/día | No es efectiva en sujetos con MMA mut0. Bioquímicamente efectiva, pero sin mejoría clínica |
| Piridoxina (B6) (Benadón®) | Cofactor de transaminasas | Atrofia girata | 300-600mg/día | Un número pequeño de sujetos responde al tratamiento |
| Riboflavina (B2) (Fórmula magistral) | Cofactor de deshidrogenasas | MADD | 100-300mg/día | Muy efectiva en sujetos con MADD, fenotipo leve o moderado |
| Tiamina (B1) (Benerva®) | Cofactor de decarboxilasas | Jarabe de arce | 300mg/día | El 20% puede responder |
CPS: carbamil fosfato sintetasa; MADD: Déficit múltiple de deshidrogenasas; MMA: metil malónico acidemia; Mut 0: falta completa de función enzimática; NAG: N acetil glutamato; NAGS: N acetil glutamato sintetasa; NCG: N carbamil glutamato.
Después de 2 horas hay que medir los valores de amonio. Si éstos aumentan o si en la primera determinación son superiores a 350μmol/l debe emplearse:
- –
Ácido carglúmico o N carbamil glutamato (Carbaglu® 200mg comprimidos dispersables) administrado por vía nasogástrica, con una dosis inicial de carga de 100 a 250mg/kg. Después deben mantenerse 100 a 200mg/kg/día, cada 6 u 8 h. Se sabe que es específico para el déficit de N-acetilglutamato sintetasa, pero también es muy útil en las hiperamonemias debidas a acidemias orgánicas7, en trastornos de la betaoxidación y en otras enfermedades de causa no aclarada. Por eso, debe usarse lo más pronto posible a fin de evitar una actuación más intensiva con medidas dialíticas.
Después de 4h se realiza un control de los valores de amonio, si estos son de 350μmol/l o más, deben iniciarse medidas físicas para depurarlo.
Medidas dialíticas:Las medidas dialíticas más indicadas son las siguientes:
- 1.
La hemodiafiltración venovenosa continua. Los métodos que se utilizan son los siguientes:
- ○
El método ideal consiste en la punción de vena femoral con catéter de doble luz de 5 a 7 fr. Es conveniente invertir los flujos de las luces en el RN y que se asuma cierta recirculación para evitar que al succionar por una luz proximal se ocluya con la pared del vaso.
- ○
Alternativas:
- •
catéter más fino de 4 fr en cada vena femoral;
- •
acceso a vena femoral por disección, si no es posible realizarlo por punción,
- •
catéter de 7 fr en vena umbilical, pero no como primera opción ya que el catéter ha de ser lo más corto posible y debe pasar por la vena porta, caso contrario, no habrá suficiente flujo de sangre.
- •
- ○
- 2.
La hemodiálisis es el tratamiento más eficaz para eliminar el amonio, pero presenta dificultades técnicas y mala tolerancia hemodinámica en RN de menos de 5kg.
Otras medidas dialíticas pueden ser las siguientes:
- 1.
La hemodiafiltración arteriovenosa continua es eficaz pero se prefiere la hemodiafiltración venovenosa.
- 2.
La diálisis peritoneal es poco eficaz pues a pesar de administrarse con altos volúmenes horarios y soluciones hipertónicas consigue un aclaramiento máximo de amonio de 10ml/min/m2.
- 3.
La ECMO (Extra-corporeal membrane oxygenation ‘oxigenación por membrana extracorpórea’) con hemodiálisis es útil en las hiperamonemias graves. La ECMO con hemofiltración es muy útil sobre todo si las cifras de amonio son muy elevadas (>1.000μmol/l).
En cuanto al seguimiento, una vez normalizadas las concentraciones de amonio con las medidas terapéuticas de urgencia, el objetivo del tratamiento nutricional es aportar una cantidad de calorías suficientes para impedir el catabolismo proteico y promover el anabolismo. Los aportes pueden variar en función del diagnóstico de sospecha inicial.
Si se empleó el benzoato sódico debe retirarse la medicación una vez que las concentraciones de amonio estén normalizadas. El resto de la medicación debe mantenerse igual hasta tener una sospecha diagnóstica clara.
