





Los feocromocitomas y los paragangliomas son tumores neuroendocrinos infrecuentes en pediatría; sin embargo, estos representan el tumor endocrino más frecuente en la infancia. La mayoría son esporádicos, cobrando mayor relevancia los síndromes familiares en edad pediátrica. Los avances en el campo de la genética, en bioquímica y en técnicas de imagen han adaptado el manejo de estos tumores; de modo que el estudio bioquímico debe comenzarse ante todo diagnóstico clínico de sospecha, seguido del estudio por imagen y molecular, más aún ante la existencia de un síndrome familiar conocido.
Revisamos aspectos clínicos, diagnósticos y terapéuticos a propósito de 2 casos, presentando el segundo paciente antecedentes de neurofibromatosis tipo 1.
Pheochromocytomas and paragangliomas are rare neuroendocrine tumors in children and most of them are sporadic. However, they represent the most common endocrine tumor in childhood, and hereditary tumor syndromes are most relevant in these age. Advances in genetic, biochemistry and imaging techniques have revised the management of these tumors; thus A biochemical study should be always initiated once the clinical diagnosis is suspected, followed by imaging and molecular studies, particularly in the context of known familial disease.
The diagnostic and therapeutic features are reviewed after the presentation of two clinical cases, where the second one is a patient with type 1 Neurofibromatosis.
Los feocromocitomas y/o paragangliomas son tumores neuroendocrinos productores de catecolaminas, con incidencia en la población general de 1:100.000 pacientes/año, siendo un 10-20% pacientes pediátricos. La sintomatología clínica es variada, generalmente asociada a la secreción hormonal. En ocasiones, el diagnóstico es casual tras una prueba de imagen o en el cribado familiar realizado en ciertos síndromes1. Suelen presentarse de forma esporádica, observándose una mayor proporción de síndromes genéticos en niños. Es por ello fundamental reconocer sus síntomas, más aún en el contexto de ciertas enfermedades familiares2,3.
Exponemos 2 casos de pacientes con esta afección que han sido atendidos en nuestro centro en los últimos 5 años.
Caso 1Niña de 14 años, consulta en atención primaria por cefalea holocraneal asociada a dolor abdominal difuso de 3 meses de evolución, de mayor intensidad en los últimos 5 días. La ecografía abdominal evidencia una masa retroperitoneal en región paraaórtica izquierda, bajo los vasos renales, con vascularización Doppler intensa y contornos bien definidos.
La exploración física es normal, salvo hipertensión arterial (HTA) mantenida (160/80mmHg). Ingresa para estudio, solicitándose analítica con hemograma, bioquímica y estudio de catecolaminas en orina, mostrando aumento de la excreción de noradrenalina (571 μg/24h) y 4-OH-metoximandelato (13,2mg/24h). Se realiza una resonancia magnética (RMN) abdominal, observándose lesión nodular retroperitoneal en la región paraaórtica izquierda, con captación de gadolinio moderada. Se realiza una gammagrafía con 123-metaiodobenzilguanidina (I-123-MIBG), sin presentar captación, y RMN cerebral, que resulta normal.
Ante la orientación diagnóstica de feocromocitoma extraadrenal, se realiza exéresis quirúrgica previo bloqueo alfaadrenérgico. El estudio anatomopatológico confirma el diagnóstico de paraganglioma simpático extrarrenal. Macroscópicamente, la pieza está delimitada por una fina cápsula con área central estrellada de aspecto mixoide. Microscópicamente, muestra nidos de células con citoplasma amplio, eosinófilo, positivas inmunohistoquímicamente para S-100, cromogranina y sinaptofisina. Se realiza un estudio genético mostrando mutación del gen SDHB (fig. 1).
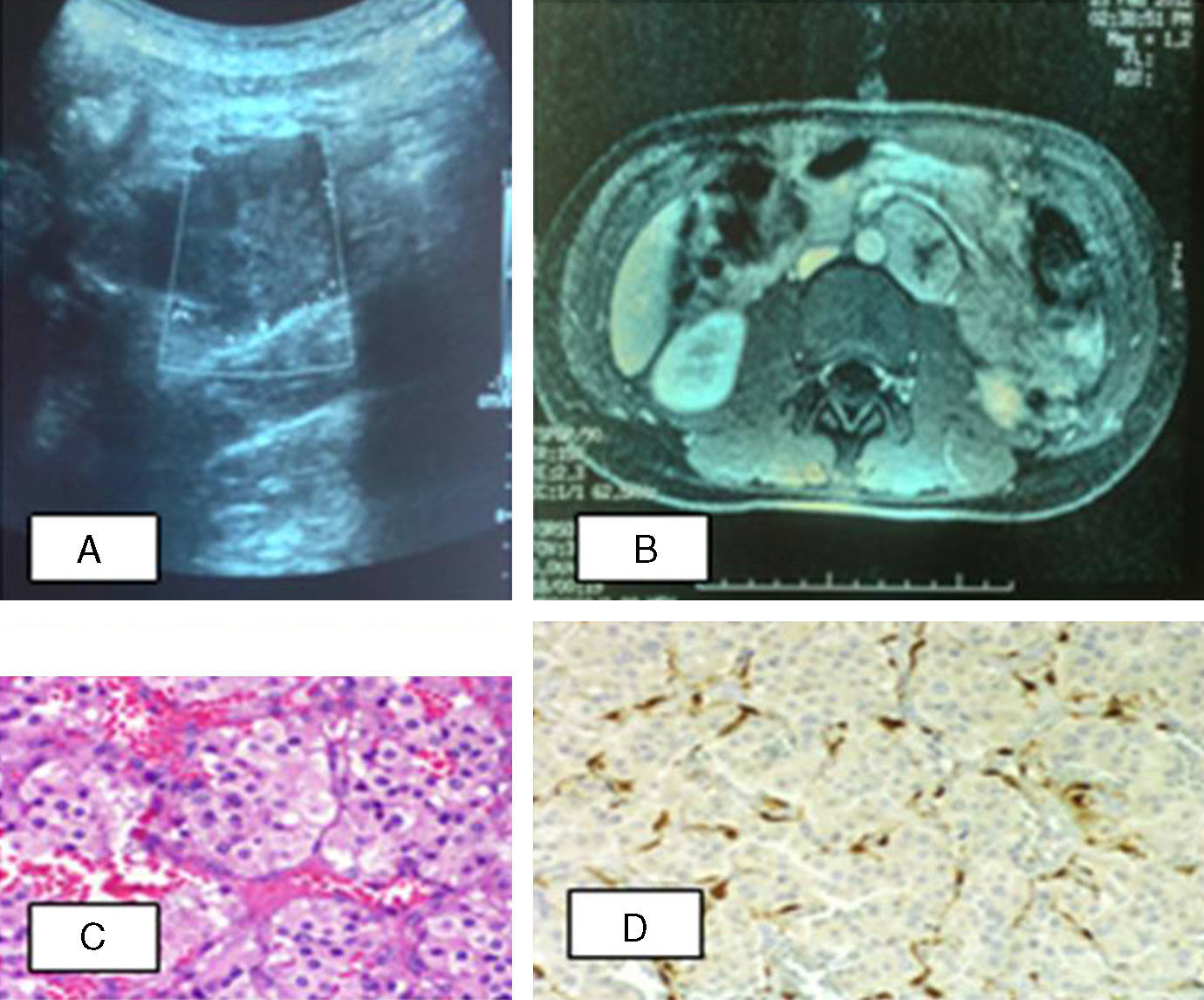
Paciente 1. A) Ecografía abdominal: en la región paraaórtica izquierda, masa de contornos bien definidos con ecogenicidad intermedia. B) RM abdominal: lesión nodular retroperitoneal, captación de contraste moderada con gadolinio. C) Tumoración constituida por nidos de células con citoplasma amplio, eosinófilo, finamente granular. D) Inmunohistoquímica: células sustentaculares positivas inmunohistoquímicamente para S-100.
Niña de 14 años con neurofibromatosis familiar tipo 1, que refiere cefalea pulsátil bitemporal, palpitaciones y vómitos desde hace 4 meses. Se solicita ecografía abdominal, objetivándose desplazamiento del riñón izquierdo por masa con vascularización escasa, en el espacio anatómico correspondiente a la glándula suprarrenal.
La exploración física es normal, salvo la presencia de neurofibromas generalizados en la piel. Ingresa para estudio con sospecha de feocromocitoma, solicitándose estudio de catecolaminas en orina. Muestra aumento de adrenalina (150 μg/24h), noradrenalina (713 μg/24h) y 4-OH-metoximandelato (24,3mg/24h). La RMN abdominal objetiva una tumoración redondeada, delimitada, de localización superoanterior al riñón izquierdo, con zonas de necrosis interna. Se solicita I-123-MIBG, presentando zona de hiperfijación a nivel de la glándula suprarrenal izquierda, compatible con el diagnóstico de presunción. La RMN cerebral resulta normal.
Tras bloqueo alfaadrenérgico, se realiza exéresis del tumor. El estudio anatomopatológico confirma el diagnóstico de feocromocitoma. Macroscópicamente, es un tumor sólido, delimitado por una fina cápsula con coloración amarillo-anaranjada. Microscópicamente, presenta nidos de células poligonales, células con citoplasma amplio eosinófilo y núcleos grandes con cromatina abierta sin necrosis. Pendiente de recibir estudio genético (fig. 2).
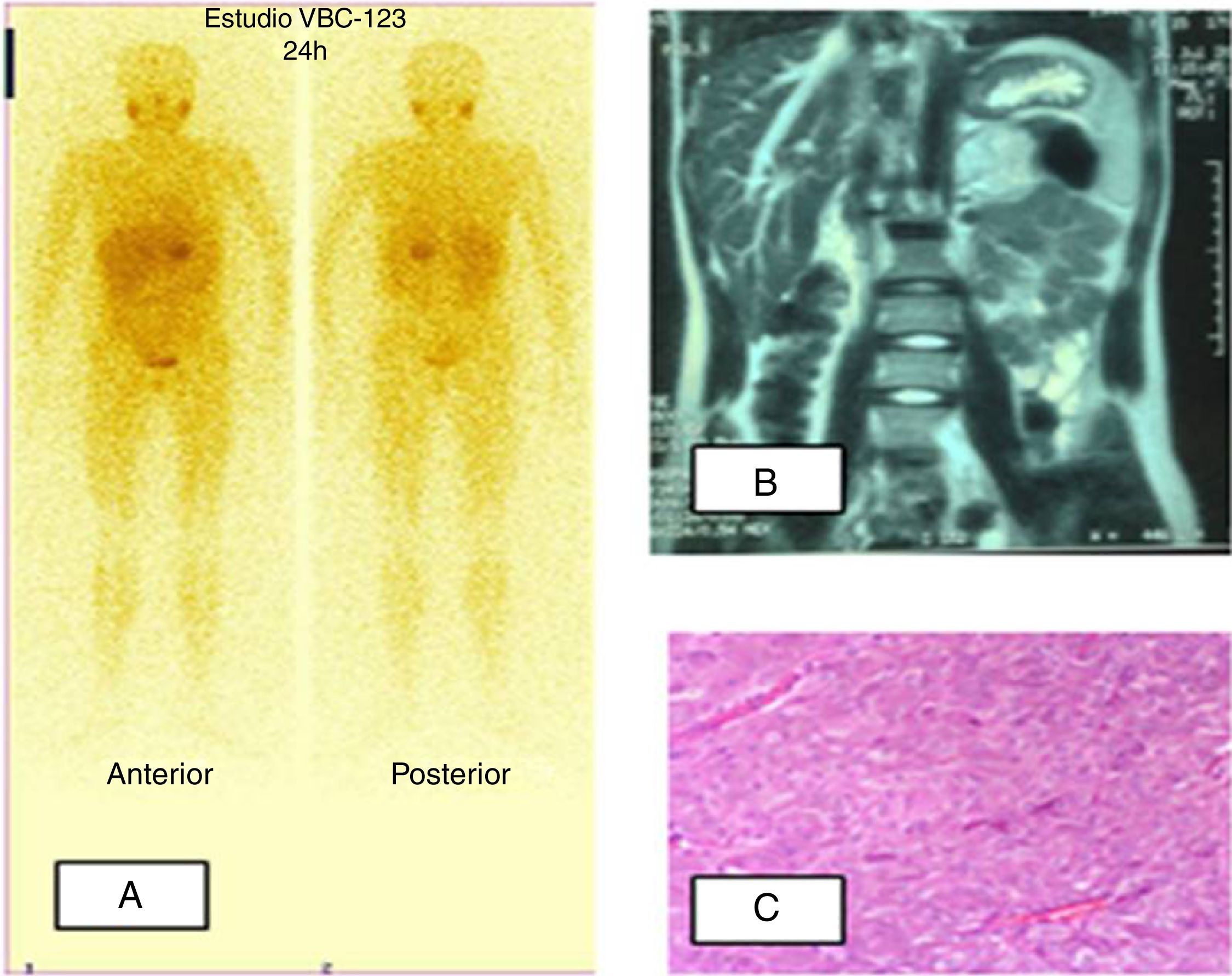
Paciente 2. A) Gammagrafía MIBG I123: hiperfijación a nivel de suprarrenal izquierda. B) RM abdominal: tumoración redondeada en localización anterior y superior al riñón izquierdo, intensidad heterogénea con hiperintensidad en T2. C) Tumoración constituida por nidos de células poligonales rodeados por finos capilares. Las células muestran citoplasma amplio eosinófilo y granular. El núcleo es grande con cromatina abierta y nucléolo patente.
El feocromocitoma y/o paraganglioma es un tumor neuroendrocrino derivado de las células cromafines de la cresta neural, denominándose feocromocitoma si asienta sobre la médula suprarrenal y paraganglioma si asienta en paraganglios simpáticos y parasimpáticos, generalmente en retroperitoneo y tórax2. Pese a que muchos autores utilizan indistintamente el término feocromocitoma para referirse a ambos, es necesario saber que existe una clara distinción en ellos, tanto por el riesgo de malignización como por el estudio genético.
Aunque la incidencia de feocromocitomas es baja, el 20% corresponde a pacientes pediátricos4, tratándose del tumor endocrino más frecuente de la infancia5. Ciertas características epidemiológicas difieren en la población infantil de la adulta: existe mayor prevalencia en el sexo masculino, mayor porcentaje de tumores bilaterales y paragangliomas3-6 y, recientemente, se ha visto que hasta en un 40% de los niños existe mutación genética conocida, aumentando hasta el 70% el porcentaje de feocromocitomas hereditarios en menores de 10 años7,8.
La sintomatología clínica es muy diversa (tabla 1), principalmente debida a la excreción de catecolaminas. La tríada clásica de cefalea, sudoración y taquicardia junto con la HTA son así signos de sobreactividad simpática. En las series pediátricas, la HTA se encuentra en un 60-90% de los casos, normalmente mantenida y no en forma de crisis hipertensivas, como ocurre en adultos3,4, y síntomas como sudoración, vómitos y poliuria son muy frecuentes7,9. Algunos pacientes permanecen asintomáticos, realizándose el diagnóstico tras una prueba de imagen de manera fortuita. Por ello, aunque sea baja la incidencia de presentar un feocromocitoma ante un incidentaloma o paciente hipertenso10,11, este debe siempre descartarse.
Manifestaciones clínicas descritas en feocromocitoma y/o paraganglioma
| Manifestaciones frecuentes | Otras |
| Hipertensión arterialCefaleaSudoración profusaTaquicardia y palpitacionesNáuseas y vómitosVisión borrosaPérdida de pesoPalidezSudoración acraTemblorDolor epigástricoDolor lumbarCrisis de ansiedad | Trastorno de conductaMalestar generalEstreñimientoDolor epigástricoDolor lumbarHipotensión ortostáticaFiebreConvulsionesAlteraciones endocrinológicasHiperglucemiaDiarrea (secreción de VIP)Poliuria (secreción inadecuada de ADH)Síndrome de Cushing (secreción de CRH/ACTH)Gigantismo (secreción GHRH)Hipercalcemia (estimulación PTH)Alteraciones cardiovascularesMiocarditisAccidentes cerebrovascularesInfarto agudo de miocardioEdema pulmonar |
La mayoría de los feocromocitomas son esporádicos, cobrando los síndromes familiares especial relevancia en edad pediátrica por su mayor frecuencia. En la tabla 2 describimos las principales características de los síndromes más representativos. Existe una importante asociación entre estos tumores aparentemente esporádicos y mutaciones reconocibles en líneas germinales del ADN en niños3,8,12, reforzando así la idea de la importancia del estudio genético ante el hallazgo de dicho tumor.
Síndromes y asociaciones familiares con feocromocitomas y/o paragangliomas
| PrevalenciaHerencia | Gen | Características clínicas generales | Características feocromocitoma,paraganglioma | |
| MEN 2MEN 2AMEN 2B | 1:40.000AD | RET (10q11.2)RET (10q11.2) | Carcinoma medular de tiroidesFeocromocitoma (50%)Adenoma/hiperplasia de paratiroides (raro)Carcinoma medular de tiroidesFeocromocitoma (50%)Ganglioneuromatosis mucosas e intestinal | 5.a-6.a década de la vidaMayoría multifocales y extradrenalesMenos del 5% malignos |
| NF1 | 1: 30.000-50.000AD | NF1 (17q11.2) | NeurofibromasManchas café con lecheNódulos de LishEscoliosis y displasias vertebralesFeocromocitoma (5%)Gliomas ópticosTumores malignos de nervios periféricosVasculopatíaEstenosis de arteria renal | 4.a década de la vidaBenigno (90%)Unilateral (75%) |
| VHL tipo 2 | 2-3:100.000AD | VHL (3p26-p25) | Hemangioblastomas en SNC y retinaFeocromocitomaTumores de saco endolinfáticoQuistes hepáticosAdenoma quístico en epidídimo y en ligamento anchoCarcinoma renalCistoadenomas páncreas | 3.a década de la vida50% bilaterales o multifocales5% malignos |
| PGLPGL1PGL2PGL3PGL4 | SDHD (11q23)SDH5, akaSDHAF2(11q13.1)SDHC (1q21)SDHB (1p36.1-p35) | FeocromocitomasParagangliomas (cabeza y cuello)Paragangliomas (cabeza y cuello)Paragangliomas (cabeza y cuello)FeocromocitomasParagangliomas (abdominal) | Mutaciones SDHD tienen impintig maternoMutaciones SDHB presentan mayor frecuencia de enfermedad metastásica |
MEN: neoplasia endocrina múltiple; NF1: neurofibromatosis tipo 1; PGL: síndrome de paraganglioma familiar VHL: síndrome de von Hippel-Lindau.
La sospecha clínica debe seguirse del diagnóstico bioquímico, debiéndose realizar en todo paciente con sintomatología compatible, ante incidentaloma abdominal y en pacientes con predisposición hereditaria conocida, siendo el fin demostrar un aumento de catacolaminas y de sus metabolitos en plasma y orina2,13-15. El diagnóstico bioquímico en niños requiere de intervalos de referencia según la edad14 y es esencial prevenir posibles causas de falsos positivos, como la ingesta de alimentos estimulantes, la actividad física o ciertos medicamentos.
Tras el estudio bioquímico se realiza el diagnóstico por imagen. No obstante, en familias con riesgo de desarrollo tumoral debe realizarse independientemente del resultado de las catecolaminas16. La ecografía, de fácil acceso y la primera a realizar en un paciente con clínica de sospecha, presenta baja sensibilidad. Por ello, se precisa de técnicas con sensibilidad elevada, como la tomografía computarizada y/o la RMN. Sin embargo, esta disminuye en tumores extraadrenales, metastásicos o recurrentes, además de presentar baja especificidad. Debe realizarse, por tanto, un estudio de imagen funcional adicional con escintigrafía, siendo la I-123-MIBG la prueba diagnóstica de elección, pues presenta gran afinidad por la médula adrenal, el tejido adrenérgico nervioso y las células del feocromocitoma. En el caso de no ser diagnóstica, puesto que puede presentar limitaciones, puede plantearse la utilización de otros radiotrazadores o métodos en el diagnóstico de localización, como es el uso de la tomografía por emisión de positrones, una nueva modalidad de escintigrafía con rendimiento similar a la MIGB, con la ventaja de no precisar bloqueo tiroideo previo y rapidez de obtención de imágenes4,16.
Tras el diagnóstico, es fundamental realizar un estudio molecular dada la alta asociación genética descrita3,8.
El tratamiento curativo es la exéresis quirúrgica, consiguiéndose un 90% de remisión completa. Este conlleva elevado riesgo cardiovascular, por lo que se deben adoptar medidas prequirúrgicas para bloquear los efectos del exceso de las catecolaminas al menos 10-14 días antes de la cirugía17, más estrecha monitorización intraoperatoria para reducir dichas complicaciones. La mayor experiencia en pacientes pediátricos se tiene con fenoxibenzamina9,12,18, antagonista no competitivo de los receptores alfa 1-postsinápticos y alfa 2-presinápticos. La dosis de fenoxibenzamina se aumenta de manera progresiva hasta conseguir estabilización de los niveles de presión arterial (inicio a 0,2mg/kg/día con aumento progresivo de 0,2mg/kg cada 4 días, dosis máxima 4mg/kg/día). Nunca debe instaurarse tratamiento betabloqueante si no se ha conseguido un bloqueo alfa efectivo, al poder desencadenar una crisis hipertensiva. Previo a la intervención quirúrgica, debe descartarse la presencia de miocardiopatía dilatada, puesto que el manejo de las complicaciones cambiaría.
La técnica quirúrgica más usada es al abordaje mediante laparoscopia16, reservando la laparotomía en enfermedad invasora o metastásica. Se realizan adrenalectomías totales en pacientes con feocromocitomas esporádicos unilaterales9 y adrenalectomía subtotal si el tamaño es menor a 5cm. En pacientes con enfermedad bilateral, se realizan adrenalectomías subtotales o adrenalectomía subtotal unilateral más adrenalectomía total contralateral con el fin de evitar la supresión adrenal19.
Como conclusiones, se debe destacar que, aunque los feocromocitomas y los paragangliomas son tumores raros en la infancia, suponen el tumor endocrino más frecuente en la edad pediátrica. Es esencial conocer la clínica indicativa de estos y, ante su sospecha, es imperativo realizar una prueba de imagen para descartar la existencia de posibles masas abdominales junto con un análisis bioquímico. Igualmente, ante un incidentaloma abdominal aislado, debe buscarse también un posible aumento de catecolaminas en el organismo. La reciente asociación de feocromocitomas y paragangliomas esporádicos a mutaciones reconocibles hace necesario realizar un estudio genético ante el hallazgo de dichos tumores.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
