



El síndrome de Smith-Lemli-Opitz (SSLO) es un error congénito del metabolismo del colesterol producido por el déficit de la enzima 7-dehidrocolesterol-reductasa, lo que condiciona generalmente la elevación de los niveles de 7-dehidrocolesterol y la disminución del colesterol durante el periodo embrionario y posnatal en plasma u otros tejidos. Se debe a variantes patogénicas bialélicas en el gen DHCR7. El fenotipo clásico se caracteriza por retraso de crecimiento de inicio prenatal, microcefalia, anomalías mayores y menores múltiples (cardiopatía, fisura palatina y genitales ambiguos fundamentalmente) y discapacidad intelectual generalmente moderada/grave1. La expresividad clínica es variable y de causa aún no aclarada2, existiendo excepcionalmente casos leves con capacidad cognitiva normal3, y otros graves con malformaciones del sistema nervioso central como la holoprosencefalia4.
Presentamos 4 casos diagnosticados pertenecientes a 2familias no relacionadas, que muestran las formas más leves y graves de la enfermedad.
Familia 1. Niña de 8 años, hija de padres sanos no consanguíneos, valorada al nacimiento por anomalías congénitas múltiples: microcefalia, rasgos dismórficos, cataratas puntiformes, fisura de paladar blando, sindactilia cutánea de 2.°-3.er dedos de pies e hipotonía con dificultades de succión. Fue diagnosticada clínicamente de SSLO (fenotipo clásico) y se solicitaron niveles de esteroles, confirmándose la elevación de 7-dehidrocolesterol (436,3 mMol/l) y disminución del colesterol total (27mg/dl). Inmediatamente empezó tratamiento con colesterol. El estudio de secuenciación del gen DHCR7 confirmó la sospecha diagnóstica al identificar las variantes patogénicas c.906C>G/c.964-1G>C.
Evolutivamente, la paciente desarrolló reflujo gastroesofágico grave; retraso psicomotor global grave (no ha adquirido deambulación ni lenguaje) y epilepsia de inicio a los 9 meses y mal control de crisis a pesar del tratamiento con levetiracetam, lamotrigina, zonegran y parampanel; hipoacusia neurosensorial moderada bilateral; infecciones respiratorias y óticas recurrentes, y obesidad.
Destacaba en sus antecedentes familiares, la interrupción de una gestación de sus padres en semana 20 por feto masculino (46, XY), sexo ecográfico femenino y holoprosencefalia. Tras el diagnóstico de la paciente, se realizó estudio genético en ADN de amniocitos, confirmándose que el feto estaba también afectado.
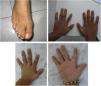
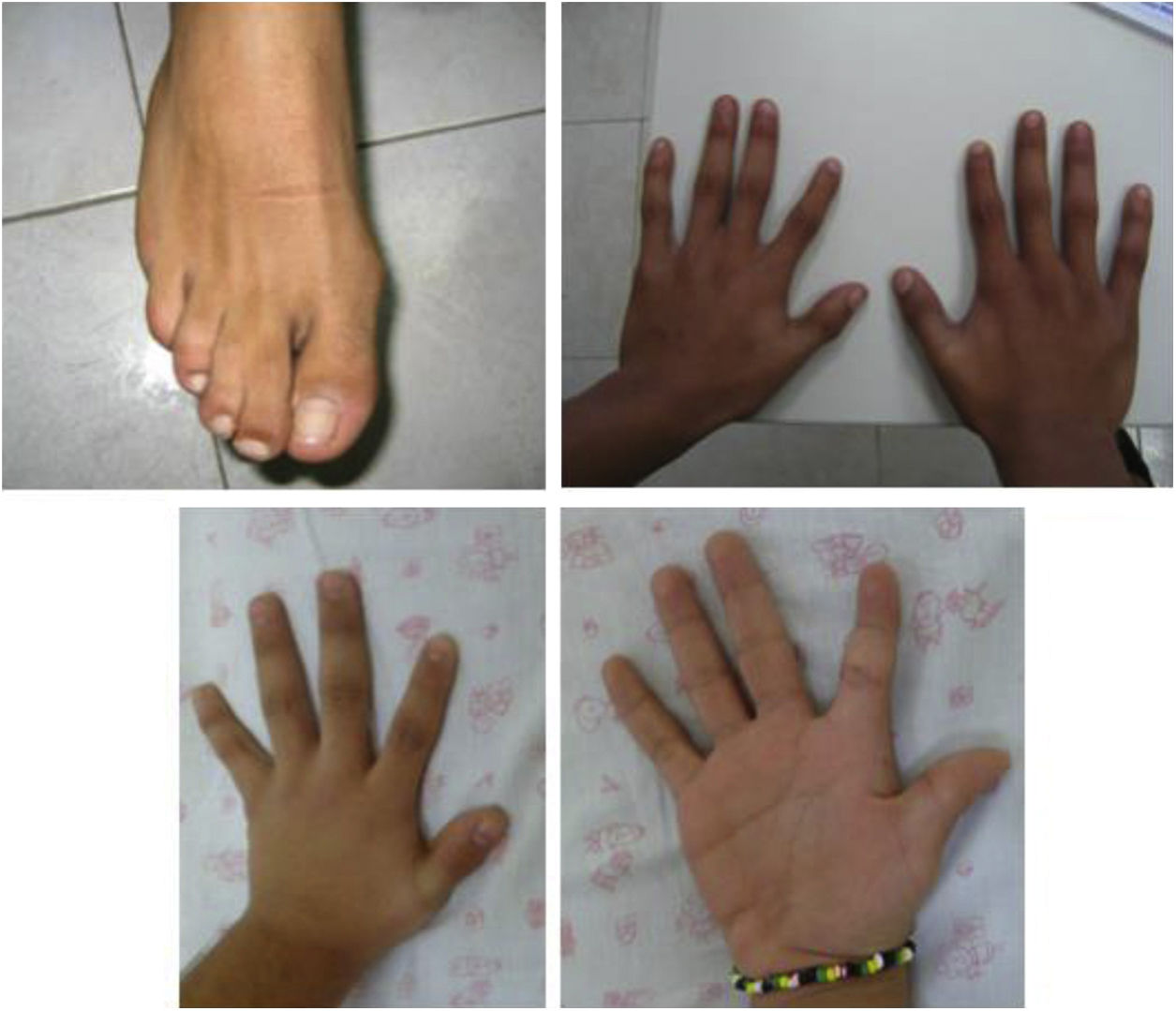
Familia 2. Varón de 22 años, hijo de padres sanos no consanguíneos, valorado a los 12 años por retraso de crecimiento de inicio prenatal, microcefalia, fisura palatina, rasgos particulares (facies alargada, puente nasal alto, narinas gruesas y leve desviación palpebral inferior), sindactilia de 2.°-3.er dedos de pies y primer dedo de manos corto e implantación baja, como se aprecia en la figura 1. Hermano de 18 años con retraso de crecimiento de inicio prenatal, úvula bífida, parecido físico y leves dificultades sociales. Desarrollo psicomotor normal en ambos. Sin cardiopatía ni enfermedad genitourinaria. Pruebas complementarias iniciales: nivel sérico de colesterol normal (145-170mg/dl), cariotipo (46,XY), MLPA 22q11 y resonancia magnética cerebral, normales. Se descartó fenilcetonuria materna. Evolutivamente, el paciente presentó trastorno de conducta moderado, impulsividad y abandono de los estudios con conductas de riesgo y consumo de drogas. Coeficiente intelectual (CI) 94 (normal-medio). El hermano ha presentado buena evolución a nivel escolar, superando objetivos, aunque con mucho esfuerzo. Relaciones sociales normales. Rechazó la valoración psicológica de CI. Precisó tubos de drenaje por otitis de repetición y cirugía por luxaciones de rótulas recurrentes. Consta en la historia clínica el nivel sérico de colesterol normal a los 6-8 años (106-125mg/dl). Estudios realizados tras nueva valoración: Array-CGH: normal; dada la edad de los pacientes (reproductiva), la sospecha de un cuadro similar con expresividad variable en ambos y al no tener una sospecha diagnóstica, se solicitó exoma clínico: detección de variantes patogénicas c.452G>A y c.1A>G en el gen DHCR7 asociadas al SSLO.
En la tabla 1 podemos ver el resumen de las manifestaciones clínicas y el genotipo de los casos.
Resumen de las características clínicas y el genotipo de los casos
| Caso 1 | Caso 2 | Caso 3 | Caso 4 | |
|---|---|---|---|---|
| Edad al diagnóstico (años) | 0 | – | 22 | 18 |
| Anomalías ecográficas prenatales | RCIU y oligoamnios | Holoprosencefalia y trastorno de la diferenciación sexual | RCIU | RCIU |
| Retraso de crecimiento posnatal | + | + | + | |
| Anomalías genitourinarias | Doble sistema excretor renal izquierdo sin dilatación | Genitales ambiguos | – | – |
| Cardiopatía | – | – | – | – |
| Sindactilia 2.° y 3.er dedos de los pies | + | + | + | |
| Fisura de paladar/úvula bífida | Fisura palatina(paladar blando) | Fisura palatina | Úvula bífida | |
| Hipoacusia | Neurosensorial moderada bilateral | |||
| Retraso psicomotor | Grave | Leve | – | |
| Discapacidad intelectual | Grave | – | – | |
| Trastorno conducta | + | + | – | |
| Nivel sérico de 7-dehidrocolesterol (μmol/l) | 436,3 | 52,32 | 10,16 | |
| Nivel sérico de colesterol total (mg/dl) | 27 | 145-170 | 106-125 | |
| Secuenciación gen DHCR7 | Variantes patogénicas c.906C>G/c.964-1G>C | Variantes patogénicas c.906C>G/c.964-1G>C | Variantes patogénicas c.452G>A/c.1A>G | Variantes patogénicas c.452G>A/c.1A>G |
La descripción de estos casos muestra de forma muy ilustrativa la expresividad altamente variable presente en pacientes con SSLO, incluso intrafamiliar. El perfil bioquímico descrito apoya su posible relación con el transporte de colesterol madre-feto durante el embarazo5.
Es importante sospechar SSLO a nivel prenatal ante la presencia de holoprosencefalia y genitales ambiguos en fetos XY, así como posnatalmente ante el retraso de crecimiento prenatal, la microcefalia, la fisura palatina y la sindactilia cutánea entre 2.°-3.er dedos de los pies, tras descartar cromosomopatías, a pesar de la normalidad en la exploración neurológica y niveles de colesterol total.
La utilidad del exoma clínico para diagnosticar casos atípicos y fenotipos leves de síndromes clásicos es clara, con las implicaciones que ello tiene no solo para el paciente al permitir plantear opciones preventivas, terapéuticas6 y un adecuado asesoramiento genético reproductivo, sino para sus familiares.
