
Los avances en el diagnóstico precoz y tratamiento han propiciado una mayor supervivencia, y en mejores condiciones, de los pacientes con enfermedades metabólicas congénitas (EMC). Estos pueden acudir a los Servicios de Urgencias Pediátricas (SUP) por motivos relacionados o no con su enfermedad. El propósito de este trabajo fue revisar las características de las visitas al SUP de estos pacientes, en un hospital de tercer nivel.
Material y métodosSe desarrolló un estudio observacional retrospectivo en el que se analizaron todas las visitas al SUP del Hospital Infantil La Paz durante los años 2011 y 2012 de pacientes con EMC. Se registraron el tipo de EMC, el motivo de consulta, el tiempo de evolución de los síntomas, la necesidad de ingreso hospitalario y la presencia de descompensación metabólica.
ResultadosEn total, fueron analizadas 107 visitas, siendo el motivo de consulta más frecuente los procesos respiratorios (30,8%). Cuando la consulta fue por vómitos, los pacientes con trastornos relacionados con las proteínas fueron los que menos tardaron en acudir al SUP. Un tercio de las visitas se siguió de ingreso, siendo la mitad de ellas por descompensación metabólica de la patología de base.
ConclusionesLos pacientes con EMC acudieron al SUP por motivos muy diversos, que en algunos casos fueron causa o consecuencia de una descompensación metabólica aguda que motivó el ingreso hospitalario. Al tratarse de enfermedades con baja prevalencia individual, resulta de interés contar con protocolos diagnóstico-terapéuticos que faciliten una atención óptima.
Advances in the early diagnosis and treatment have led to improved survival, and a better quality of life for patients with inherited metabolic disorders (IMD). They can go to the Pediatric Emergency Services (PES) for reasons unrelated to their disease. The purpose of this study was to review the characteristics of visitors to the PES of these patients in a tertiary hospital.
Material and methodsA retrospective observational study was conducted on all visits from patients with IMD to the PES of Hospital Infantil La Paz over the years 2011 and 2012. IMD type, complaint, duration of symptoms, need for hospitalization, and presence of metabolic decompensation was recorded.
ResultsA total of 107 visits were analyzed, with the most frequent reason being for consultation of respiratory processes (30.8%). When the consultation was for vomiting, patients with protein-related disorders were those who delayed less in going to PES. One third of visitors were admitted, half of them due to metabolic decompensation of the underlying pathology.
ConclusionsPatients with IMD came to PES for many different reasons, which in some cases were the cause or consequence of an acute metabolic decompensation that led to hospitalization. Being diseases with low prevalence, it would be useful to have diagnostic and therapeutic protocols in order to provide optimal care.
Las enfermedades metabólicas congénitas (EMC) son procesos hereditarios de base genética. Su prevalencia individual es baja; sin embargo, si se consideran de forma global, su frecuencia asciende aproximadamente a 1/600 recién nacidos vivos1,2. Hasta hace unos años, se asociaban muchas veces a una evolución fatal o al desarrollo de graves secuelas e importante discapacidad. Actualmente, disponemos de herramientas que permiten, cada vez en más casos su detección precoz y la aplicación de tratamientos que mejoren la calidad de vida y la evolución clínica de estos pacientes. Esto ha tenido como consecuencia una presencia cada vez mayor en las consultas de pediatría y en los servicios de urgencias, de niños diagnosticados de EMC que consultan por diversos motivos, relacionados o no con su patología de base3. Esta circunstancia se ha visto incrementada desde que contamos con programas de cribado metabólico neonatal ampliado, que permiten la supervivencia con buena calidad de vida de numerosos pacientes4-6. Los pediatras que prestan asistencia en los servicios de urgencias no deben ser ajenos a esta población, que puede requerir atención urgente a pesar de consultar por un motivo de aparentemente poca gravedad, pero que puede ser causa de descompensación metabólica de su enfermedad de base7.
El objetivo de este trabajo es revisar la frecuentación de un servicio de urgencias pediátricas (SUP) por parte de niños diagnosticados de EMC.
Pacientes y métodosSe desarrolló un estudio observacional retrospectivo en el que se analizaron las visitas al SUP del Hospital Universitario La Paz, durante los años 2011 y 2012, de pacientes diagnosticados de EMC que fueran seguidos en ese periodo por la Unidad de Nutrición Infantil y Enfermedades Metabólicas del centro. En el estudio no se incluyó a los pacientes con dislipidemias genéticas.
Durante el periodo de estudio, 58 pacientes con EMC fueron seguidos en la consulta de Nutrición y Enfermedades Metabólicas. Atendiendo a su patología de base, fueron clasificados en 4 grupos:
- 1.
Trastornos del metabolismo de los hidratos de carbono (TMHC): se incluyeron los pacientes con trastornos del metabolismo del glucógeno, trastornos del metabolismo de la galactosa, trastornos del metabolismo de la fructosa, hiperinsulinismos congénitos y defectos en el transporte intracelular de la glucosa. Total: 7 pacientes.
- 2.
Trastornos relacionados con el metabolismo de los lípidos (TML): pacientes con trastornos de la betaoxidación de los ácidos grasos, trastornos del sistema de la carnitina y trastornos de la síntesis y utilización de los cuerpos cetónicos. Total: 8 pacientes.
- 3.
Trastornos del metabolismo de las proteínas/aminoácidos (TMP/A): en este grupo se incluyó a los pacientes con hiperfenilalaninemias, trastornos del metabolismo de la tirosina, hiperglucinemia no cetósica, acidemia propiónica, acidemia metilmalónica y trastornos del transporte y metabolismo intracelular de la vitamina B12, enfermedad de la orina de jarabe de arce, metilcrotonilglicinuria, aciduria glutárica tipo i, homocistinuria clásica y trastornos del metabolismo de los aminoácidos azufrados, histidinemia y trastornos del ciclo de la urea. Total: 40 pacientes.
- 4.
Enfermedades mitocondriales (EM): pacientes con déficits únicos o combinados de la cadena respiratoria y fosforilación oxidativa, y pacientes con déficit de piruvato deshidrogenasa. Total: 3 pacientes.
Además de los anteriores, la casuística de la unidad incluye a 5 pacientes con trastornos del metabolismo de moléculas complejas, concretamente con defectos congénitos de glucosilación y enfermedades lisosomales. De ellos, solo un paciente con defecto congénito de glucosilación de proteínas tipo IA acudió al SUP en el periodo en estudio. En el estudio estadístico se incluyó a este paciente en el grupo de TMP/A.
En cada paciente se registraron el número de visitas al SUP en el periodo en estudio, el motivo de consulta, el tiempo de evolución de los síntomas, los ingresos hospitalarios desde el SUP y la presencia de descompensación metabólica asociada. Para el análisis estadístico se empleó el programa StatCalc 5.0. Se utilizó la prueba estadística de la χ2 para la comparación de variables no paramétricas. El nivel de significación estadística se definió en p<0,05.
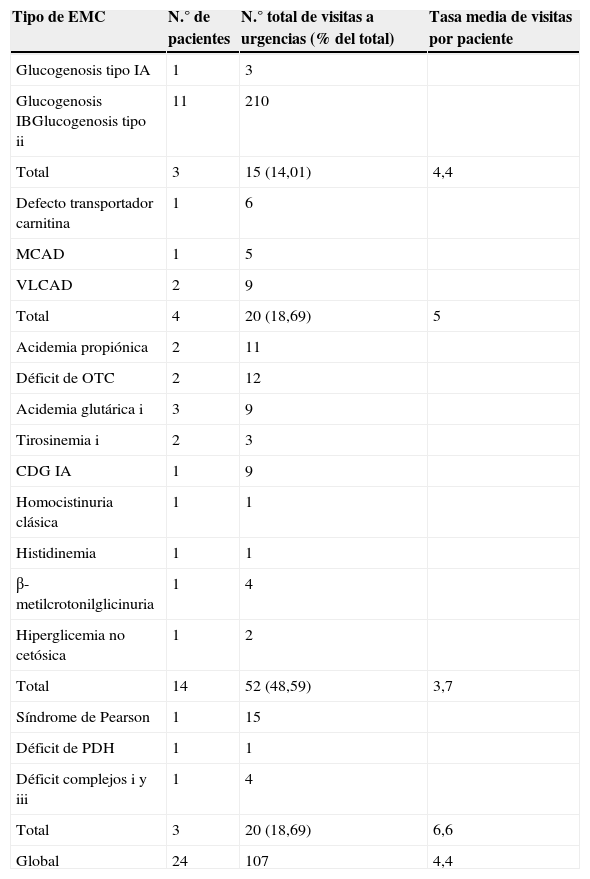
ResultadosEn total fueron incluidos 24 pacientes en nuestra revisión (tabla 1). El grupo correspondiente a los TMP/A fue el más numeroso (14 pacientes), con edades comprendidas entre los 16 años y 10 meses de la última visita de un paciente con déficit de ornitina transcarbamilasa (OTC) y los 2 meses de un lactante que ingresó en fallo hepático, siendo posteriormente diagnosticado de tirosinemia tipo 1. La edad media de este grupo de pacientes fue 11,9 años.
Pacientes con EMC atendidos en urgencias (2011-2012). Relación entre el tipo de EMC y la frecuencia de visitas a urgencias (n = 107 visitas)
| Tipo de EMC | N.° de pacientes | N.° total de visitas a urgencias (% del total) | Tasa media de visitas por paciente |
|---|---|---|---|
| Glucogenosis tipo IA | 1 | 3 | |
| Glucogenosis IBGlucogenosis tipo ii | 11 | 210 | |
| Total | 3 | 15 (14,01) | 4,4 |
| Defecto transportador carnitina | 1 | 6 | |
| MCAD | 1 | 5 | |
| VLCAD | 2 | 9 | |
| Total | 4 | 20 (18,69) | 5 |
| Acidemia propiónica | 2 | 11 | |
| Déficit de OTC | 2 | 12 | |
| Acidemia glutárica i | 3 | 9 | |
| Tirosinemia i | 2 | 3 | |
| CDG IA | 1 | 9 | |
| Homocistinuria clásica | 1 | 1 | |
| Histidinemia | 1 | 1 | |
| β-metilcrotonilglicinuria | 1 | 4 | |
| Hiperglicemia no cetósica | 1 | 2 | |
| Total | 14 | 52 (48,59) | 3,7 |
| Síndrome de Pearson | 1 | 15 | |
| Déficit de PDH | 1 | 1 | |
| Déficit complejos i y iii | 1 | 4 | |
| Total | 3 | 20 (18,69) | 6,6 |
| Global | 24 | 107 | 4,4 |
CDG IA: defecto congénito de glucosilación tipo IA; MCAD: insuficiencia de acil-CoA deshidrogenasa de cadena media; OTC: ornitina transcarbamilasa; PDH: piruvato deshidrogenasa; VLCAD: déficit de acil-CoA deshidrogenasa de cadena muy larga.
El grupo de los TML, formado por 4 pacientes, se caracterizó por ser el más homogéneo en cuanto a su edad (media: 1,2 años, mediana: 0,9 años, moda: 0,8 años).
El grupo de los TMHC lo constituyeron 3 pacientes: un adolescente de 16 años con glucogenosis tipo IA, un niño de 3 años con glucogenosis tipo IB y una niña con glucogenosis tipo ii (que fue la que generó el mayor número de visitas a urgencias, 10 en total), cuya primera visita fue con 6 meses y la última al año y 7 meses de edad.
El paciente que generó el mayor número de visitas a urgencias (15) perteneció al grupo de las EM, un niño con síndrome de Pearson cuya primera visita fue con 8 meses y la última con 2 años y 8 meses de edad. La media de edad de los pacientes con EM fue de 6 años.
Relación entre el grupo de enfermedades metabólicas congénitas y la frecuencia de visitas a urgenciasDurante el periodo de estudio, del total de 58 pacientes con EMC seguidos en la consulta de Nutrición y Enfermedades Metabólicas, 24 pacientes (41%) acudieron al SUP, generando un total de 107 visitas. El porcentaje más elevado de las mismas, con un 48,5% (52/107), correspondió a pacientes con TMP/A; sin embargo, tras analizar el número de consultas por paciente, se observó que los pacientes con una tasa media más elevada fueron los pertenecientes al grupo de las enfermedades mitocondriales (6,6 visitas por paciente) seguido de los pacientes del grupo de los TML (5 visitas), TMHC (4,4 visitas) y TMP/A (3,7 visitas) (tabla 1).
Si tenemos en cuenta al total de pacientes seguidos en consulta durante el periodo de estudio, la frecuentación es semejante (EM 6,6 visitas por paciente. TML: 2,5. TMHC: 2,1. TMP/A: 1,3 visitas por paciente).
El turno con más frecuentación fue el de tarde (39% de las visitas), seguido de la mañana (33,6%) y, por último, la noche (27,1%). En relación con el tiempo medio de evolución de la EMC de base, fue similar en los TMHC, TMP/A y EM (42, 32 y 29 meses, respectivamente).
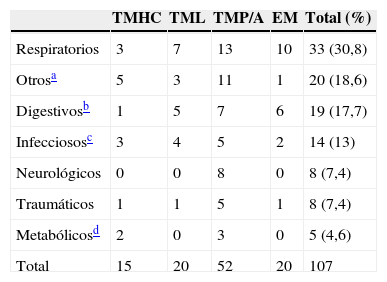
Análisis de los motivos de consultaEl motivo de consulta más frecuente fueron los procesos respiratorios (30,8%). Este grupo incluyó todas las visitas motivadas por sintomatología respiratoria, asociada o no a fiebre, rechazo de tomas/vómitos y dificultad respiratoria (tabla 2). La mayoría de estos pacientes no precisaron medidas terapéuticas específicas por su enfermedad de base, manejándose de forma similar a los que no tienen EMC.
Motivos de consulta en urgencias de los pacientes con EMC (N.° de visitas)
| TMHC | TML | TMP/A | EM | Total (%) | |
|---|---|---|---|---|---|
| Respiratorios | 3 | 7 | 13 | 10 | 33 (30,8) |
| Otrosa | 5 | 3 | 11 | 1 | 20 (18,6) |
| Digestivosb | 1 | 5 | 7 | 6 | 19 (17,7) |
| Infecciososc | 3 | 4 | 5 | 2 | 14 (13) |
| Neurológicos | 0 | 0 | 8 | 0 | 8 (7,4) |
| Traumáticos | 1 | 1 | 5 | 1 | 8 (7,4) |
| Metabólicosd | 2 | 0 | 3 | 0 | 5 (4,6) |
| Total | 15 | 20 | 52 | 20 | 107 |
EM: enfermedades mitocondriales; TMHC: trastornos del metabolismo de los hidratos de carbono: TML: trastornos del metabolismo de los lípidos; TMP/A: trastornos del metabolismo de las proteínas/aminoácidos.
Se incluyeron en este grupo los siguientes motivos de consulta: llanto/irritabilidad, dermatológicos, astenia/decaimiento, problemas relacionados con aparato locomotor de origen no traumático, problemas dispositivos de alimentación, así como otros motivos de consulta: ajustes de tratamiento y un paciente que acudió por hallazgo casual de tumoración en suelo de la boca.
El segundo grupo más frecuente de motivos de consulta lo constituyeron los síntomas de carácter digestivo. Entre ellos, la causa más frecuente de visita al SUP fueron los vómitos aislados (11,21%), observándose que el tiempo de evolución de los mismos fue inferior en los pacientes del grupo de los TMP/A (tiempo medio de evolución de los vómitos de 8,4 h) en relación con el resto de los otros 3 grupos (TMHC: 60 h, TML: 48 h, EM: 69,6 h).
Otros motivos de consultaAparte de los motivos de origen traumatológico (8 visitas), se contabilizó una gran variedad de motivos de consulta (llanto/irritabilidad, dermatológicos, astenia/decaimiento, problemas relacionados con el aparato locomotor de origen no traumático, sintomatología miccional, dudas sobre el tratamiento, etc.), que contabilizaron un total de 20 visitas. El motivo de consulta más frecuente dentro de este grupo fueron los problemas de los dispositivos de alimentación (9/20, 45%).
Ninguno de los pacientes que consultó por patología del aparato locomotor, problemas dermatológicos y sintomatología miccional precisó medidas específicas en función de su patología metabólica de base.
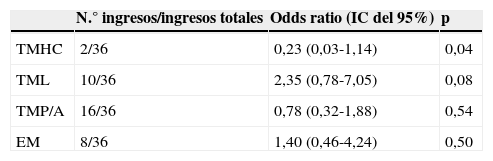
Ingresos desde el Servicio de Urgencias Pediátricas de los pacientes con enfermedades metabólicas congénitasEl 33,64% de las consultas (36/107) se siguió de ingreso hospitalario. En cuanto a la asociación entre el tipo de EMC y el riesgo de ingreso, encontramos un resultado estadísticamente significativo en el grupo de los TMHC (tabla 3).
Riesgo de ingreso según el tipo de EMC
| N.° ingresos/ingresos totales | Odds ratio (IC del 95%) | p | |
|---|---|---|---|
| TMHC | 2/36 | 0,23 (0,03-1,14) | 0,04 |
| TML | 10/36 | 2,35 (0,78-7,05) | 0,08 |
| TMP/A | 16/36 | 0,78 (0,32-1,88) | 0,54 |
| EM | 8/36 | 1,40 (0,46-4,24) | 0,50 |
EM: enfermedades mitocondriales; TMHC: trastornos del metabolismo de los hidratos de carbono; TML: trastornos del metabolismo de los lípidos; TMP/A: trastornos del metabolismo de las proteínas/aminoácidos.
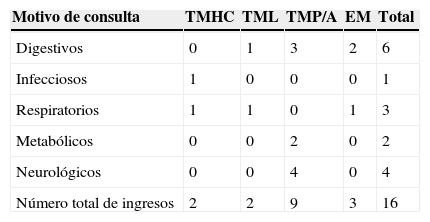
De los 36 ingresos, 16 (44,4%) fueron motivados por descompensación metabólica de la enfermedad de base (tabla 4). El motivo de consulta que con mayor frecuencia se asoció a descompensación fueron los procesos digestivos (6/16 visitas: 37,5%), concretamente los vómitos aislados, seguido de los cuadros neurológicos (4/16 visitas: 25%) y respiratorios (3/16 visitas: 18,7%). El tiempo medio de espera antes de consultar en urgencias en los pacientes que ingresaron por descompensación metabólica fue de 27,13 h.
Número total de ingresos por descompensación metabólica en función de motivo de consulta inicial en urgencias
| Motivo de consulta | TMHC | TML | TMP/A | EM | Total |
|---|---|---|---|---|---|
| Digestivos | 0 | 1 | 3 | 2 | 6 |
| Infecciosos | 1 | 0 | 0 | 0 | 1 |
| Respiratorios | 1 | 1 | 0 | 1 | 3 |
| Metabólicos | 0 | 0 | 2 | 0 | 2 |
| Neurológicos | 0 | 0 | 4 | 0 | 4 |
| Número total de ingresos | 2 | 2 | 9 | 3 | 16 |
EM: enfermedades mitocondriales; TMHC: trastornos del metabolismo de los hidratos de carbono; TML: trastornos del metabolismo de los lípidos; TMP/A: trastornos del metabolismo de las proteínas/aminoácidos.
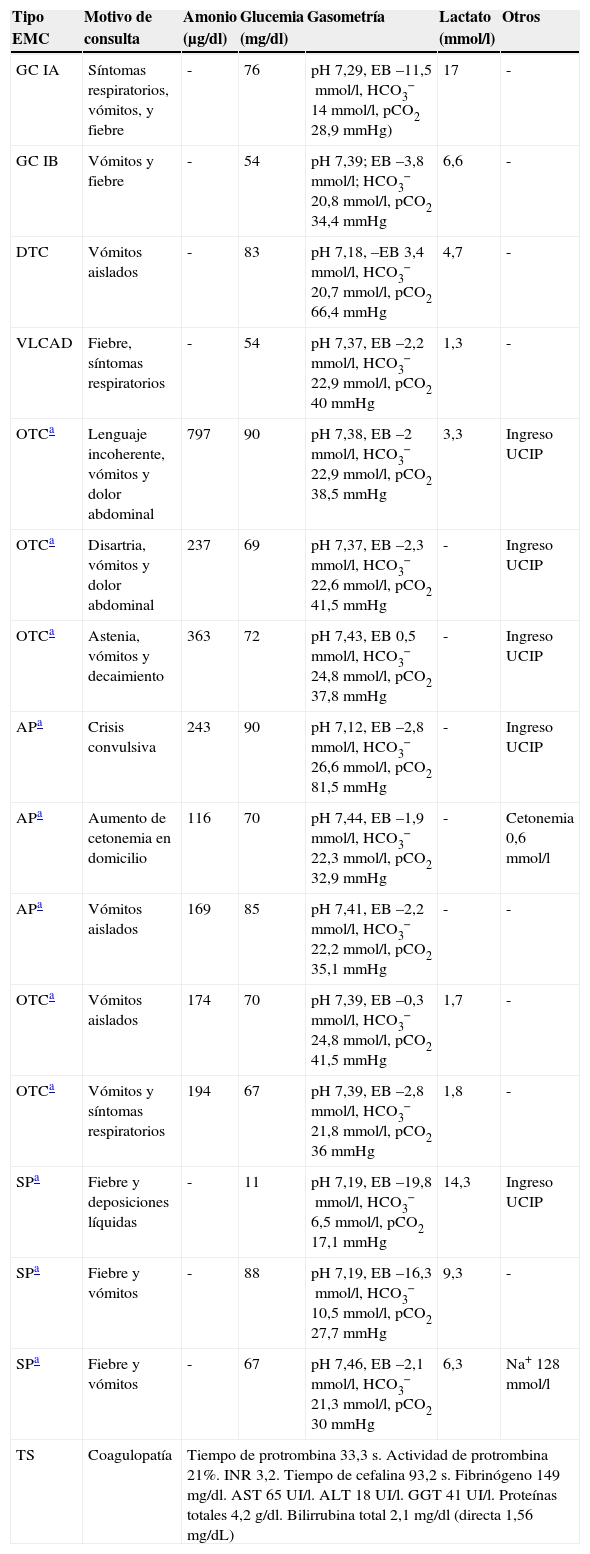
Los pacientes que ingresaron con descompensación metabólica presentaron características diferentes en función de su patología de base (tabla 5):
- 1.
TMHC: 2 ingresos. El primero, un paciente con una glucogenosis tipo IA, que consultó por fiebre de corta evolución acompañada de síntomas respiratorios y vómitos; asociando mal control de glucemias en su domicilio los días previos. En urgencias, los niveles de glucosa resultaron normales asociando acidosis metabólica. El segundo presentaba una glucogenosis tipo IB y también consultó por un cuadro febril con vómitos; sus padres no habían detectado hipoglucemias en su domicilio. En el SUP se objetivó hipoglucemia de 54mg/dl, acompañada de hiperlactadicemia y acidosis metabólica compensada. Se instauró fluidoterapia y se decidió su ingreso ante la falta de tolerancia oral adecuada.
- 2.
TML: 2 ingresos. El primero se trató de una lactante de 28 días de vida con un defecto de acil-coA deshidrogenasa de cadena muy larga (VLCAD), cuyo motivo de consulta fue la presencia de fiebre y síntomas respiratorios, con dificultades para la alimentación.
Su glucemia inicial fue de 54mg/dl, iniciando fluidoterapia por vía intravenosa con aportes suplementarios de glucosa (7mg/kg/min). La gasometría y el lactato fueron normales. El segundo paciente presentaba un déficit del transportador de carnitina y acudió por vómitos aislados. En urgencias, se objetivó hiperlactacidemia, acidosis y glucemia normal.
- 3.
TMP/A: 9 ingresos, 8 de los cuales motivados por la presencia de hiperamoniemia (rango de normalidad para mayores de un mes de nuestro laboratorio: 15-45μg/dl); correspondientes a 2 pacientes con déficit de ornitina transcarbamilasa y una con acidemia propiónica (ninguno de ellos de menos de un mes de vida). El ingreso restante se correspondió con un lactante de 2 meses de vida que fue trasladado a nuestro hospital para estudio de coagulopatía. El motivo inicial de consulta fue un episodio de posible atragantamiento asociado a desconexión del medio e hipotonía, lo que motivó la realización de la analítica inicial. A su llegada a urgencias, la exploración neurológica resultó normal, objetivándose una hepatomegalia de 1-2cm. Se realizó una segunda analítica que confirmó la coagulopatía y el paciente ingresó para estudio. Posteriormente, se confirmó el diagnóstico de tirosinemia tipo 1.
- 4.
EM: 3 ingresos, que correspondieron todos ellos a un mismo paciente con síndrome de Pearson. En el primero, el motivo de consulta fueron los vómitos aislados y el paciente presentó en urgencias una hiponatremia de 128mmol/l asociada a acidosis láctica con alcalosis respiratoria compensadora. En el segundo, la descompensación consistió en una acidosis metabólica en el contexto de una infección respiratoria con vómitos asociados, y el tercer ingreso fue debido al desarrollo de acidosis láctica intensa junto con hipoglucemia grave (11mg/dl) secundarias a gastroenteritis.
Ingresos por descompensación metabólica de los pacientes con EMC atendidos en urgencias (2011-2012). Descripción del tipo de descompensación
| Tipo EMC | Motivo de consulta | Amonio (μg/dl) | Glucemia (mg/dl) | Gasometría | Lactato (mmol/l) | Otros |
|---|---|---|---|---|---|---|
| GC IA | Síntomas respiratorios, vómitos, y fiebre | - | 76 | pH 7,29, EB –11,5mmol/l, HCO3− 14mmol/l, pCO2 28,9mmHg) | 17 | - |
| GC IB | Vómitos y fiebre | - | 54 | pH 7,39; EB –3,8mmol/l; HCO3− 20,8mmol/l, pCO2 34,4mmHg | 6,6 | - |
| DTC | Vómitos aislados | - | 83 | pH 7,18, –EB 3,4mmol/l, HCO3− 20,7mmol/l, pCO2 66,4mmHg | 4,7 | - |
| VLCAD | Fiebre, síntomas respiratorios | - | 54 | pH 7,37, EB –2,2mmol/l, HCO3− 22,9mmol/l, pCO2 40mmHg | 1,3 | - |
| OTCa | Lenguaje incoherente, vómitos y dolor abdominal | 797 | 90 | pH 7,38, EB –2mmol/l, HCO3− 22,9mmol/l, pCO2 38,5mmHg | 3,3 | Ingreso UCIP |
| OTCa | Disartria, vómitos y dolor abdominal | 237 | 69 | pH 7,37, EB –2,3mmol/l, HCO3− 22,6mmol/l, pCO2 41,5mmHg | - | Ingreso UCIP |
| OTCa | Astenia, vómitos y decaimiento | 363 | 72 | pH 7,43, EB 0,5mmol/l, HCO3− 24,8mmol/l, pCO2 37,8mmHg | - | Ingreso UCIP |
| APa | Crisis convulsiva | 243 | 90 | pH 7,12, EB –2,8mmol/l, HCO3− 26,6mmol/l, pCO2 81,5mmHg | - | Ingreso UCIP |
| APa | Aumento de cetonemia en domicilio | 116 | 70 | pH 7,44, EB –1,9mmol/l, HCO3− 22,3mmol/l, pCO2 32,9mmHg | - | Cetonemia 0,6mmol/l |
| APa | Vómitos aislados | 169 | 85 | pH 7,41, EB –2,2mmol/l, HCO3− 22,2mmol/l, pCO2 35,1mmHg | - | - |
| OTCa | Vómitos aislados | 174 | 70 | pH 7,39, EB –0,3mmol/l, HCO3− 24,8mmol/l, pCO2 41,5mmHg | 1,7 | - |
| OTCa | Vómitos y síntomas respiratorios | 194 | 67 | pH 7,39, EB –2,8mmol/l, HCO3− 21,8mmol/l, pCO2 36mmHg | 1,8 | - |
| SPa | Fiebre y deposiciones líquidas | - | 11 | pH 7,19, EB –19,8mmol/l, HCO3− 6,5mmol/l, pCO2 17,1mmHg | 14,3 | Ingreso UCIP |
| SPa | Fiebre y vómitos | - | 88 | pH 7,19, EB –16,3mmol/l, HCO3− 10,5mmol/l, pCO2 27,7mmHg | 9,3 | - |
| SPa | Fiebre y vómitos | - | 67 | pH 7,46, EB –2,1mmol/l, HCO3− 21,3mmol/l, pCO2 30mmHg | 6,3 | Na+ 128mmol/l |
| TS | Coagulopatía | Tiempo de protrombina 33,3 s. Actividad de protrombina 21%. INR 3,2. Tiempo de cefalina 93,2 s. Fibrinógeno 149mg/dl. AST 65 UI/l. ALT 18 UI/l. GGT 41 UI/l. Proteínas totales 4,2g/dl. Bilirrubina total 2,1mg/dl (directa 1,56mg/dL) | ||||
ALT: alanina aminotransferasa; AP: acidemia propiónica; AST: aspartato aminotransferasa; DM: descompensación metabólica; DTC: defecto en el transportador de la carnitina; EB: exceso de bases; GC IA: glucogenosis tipo IA; GC IB: glucogenosis tipo IB; GGT: gamma glutamil transpeptidasa; INR: Relación Normalizada Internacional; OTC: deficiencia de ornitina-transcarbamilasa; SP: síndrome de Pearson; TS: tirosinemia tipo 1; UCIP: Unidad de cuidados intensivos pediátricos; VLCAD: defecto de acil-coA deshidrogenasa de cadena muy larga.
Dentro del grupo de ingresos no asociados a descompensación metabólica (20/36: 55,5%), el 60% (12/20) se debió a procesos febriles y digestivos correspondientes a 4 visitas de pacientes con EM y 4 con TML. En el 41,6% de estos casos (5/12) el motivo de ingreso fue el riesgo de descompensación metabólica por falta de tolerancia oral. Del resto, las afecciones respiratorias supusieron un 25% de los ingresos (5/20) y el ingreso de estos pacientes no guardó relación con el riesgo de descompensación: 2 pacientes con neumonía, que ingresaron por la necesidad de tratamiento antibiótico intravenoso (MCAD y síndrome de Pearson) y 3 pacientes con insuficiencia respiratoria aguda (acidemia propiónica, homocistinuria clásica y acidemia glutárica) por necesidad de oxigenoterapia.
DiscusiónNo se han encontrado diferencias significativas que relacionen el tipo de EMC con un mayor riesgo de ingreso. Aunque en el caso de los TMHC la asociación resulta estadísticamente significativa, este resultado tiene poca importancia desde el punto de vista clínico porque la odds ratio para ser un estudio observacional es muy pequeña y además el intervalo de confianza roza el valor nulo.
El paciente con EMC suele acudir a urgencias ante mínimos cambios que aparezcan de forma aguda en su estado general, ante la posibilidad de que el cuadro derive en una descompensación metabólica8. En este caso, el manejo en urgencias difiere notablemente del niño sin EMC, pues el objetivo del pediatra es prevenir o detectar de manera precoz una posible descompensación y tratar las alteraciones que puedan comprometer la vida del paciente9-11. En esta serie, la mayoría de los pacientes con descompensación presentaron una o más de las siguientes alteraciones bioquímicas: trastornos del equilibrio ácido-base o la coagulación, hiperamoniemia, hipoglucemia y alteraciones electrolíticas12,13. En ocasiones, resulta difícil diferenciar si la clínica inicial del paciente es secundaria a la propia descompensación o al proceso intercurrente asociado.
La hiperamoniemia fue observada en pacientes del grupo de los TMP/A, concretamente en trastornos del ciclo de la urea14–17 y acidemias orgánicas17,18. En el caso de pacientes con trastornos del ciclo de la urea, estos consultaron por sintomatología neurológica o vómitos aislados (ambas manifestaciones pueden ser síntomas de elevación del amonio en sangre), mientras que en los pacientes con acidemias orgánicas fue la elevación de la cetonemia lo que motivó la decisión de acudir al SUP, con independencia de la intensidad de los síntomas acompañantes (fundamentalmente, síntomas catarrales y vómitos aislados). La hiperamoniemia puede ir acompañada de alcalosis respiratoria debido al efecto estimulante del amonio a nivel del centro respiratorio del tronco cerebral, por lo que no se debe infravalorar el hallazgo exploratorio de taquipnea en un paciente con EMC19.
La hipoglucemia puede observarse en EMC de distinta índole y el retraso en el tratamiento de urgencia puede originar daño cerebral permanente20,21. En nuestra serie, hubo 2 ingresos motivados por hipoglucemia en el grupo de los TMHC, ambos en niños con glucogenosis tipo i. En esta entidad, el defecto del complejo glucosa-6-fosfatasa condiciona una incapacidad para obtener glucosa a partir de la glucogenólisis y gluconeogénesis, activándose las vías de catabolismo de los lípidos. Los hallazgos bioquímicos incluyen acidosis metabólica, hiperlactacidemia y dislipidemia. Los pacientes con glucogenosis tipo IB pueden asociar también neutropenia que predispone a las infecciones bacterianas graves; en nuestra serie, el recuento de neutrófilos en el paciente con glucogenosis IB fue normal (1.510/μL, con cifra de leucocitos totales de 5.000/μL).
Los otros episodios de hipoglucemia fueron observados en un paciente con síndrome de Pearson y en un paciente con VLCAD. El síndrome de Pearson es una infrecuente citopatía mitocondrial multiorgánica en la que la causa de las hipoglucemias se relaciona con su afectación hepática. En la VLCAD los episodios de hipoglucemia hipocetósica22 pueden desencadenarse por el ayuno, el ejercicio físico prolongado o una enfermedad intercurrente. Igualmente, los pacientes con defectos en el metabolismo de la carnitina también presentan riesgo de desarrollar hipoglucemia hipocetósica secundaria al fallo en la betaoxidación de los ácidos grasos de cadena larga. Por tanto, en ambas enfermedades está indicada la prevención de la hipoglucemia realizando comidas frecuentes ricas en hidratos de carbono complejos y evitando los periodos de ayuno prolongado.
En relación con la acidosis metabólica, es un hallazgo común en niños enfermos y es clave en la investigación de la misma determinar la presencia o ausencia de cetonas y el anión gap. La presencia de acidosis con gap normal debe ser considerada como indicador de pérdida de bicarbonato, ya sea de causa renal o intestinal9,23. Sin embargo, en las EMC, suele existir un aumento de anión gap, que indica presencia de metabolitos anormales como cetoácidos, ácido láctico o el ácido orgánico que no puede ser metabolizado8. La acidosis metabólica puede estar presente en los trastornos del metabolismo de los aminoácidos, del metabolismo del piruvato, en enfermedades mitocondriales y también en los trastornos del metabolismo de hidratos de carbono17,24,25. La acidosis láctica es un hallazgo frecuente en los trastornos mitocondriales, trastornos por almacenamiento de glucógeno y los trastornos de la gluconeogénesis24,25, entre otros.
Durante nuestro periodo de estudio, ningún caso de EMC fue diagnosticado a partir de una primera visita a nuestro SUP, exceptuando el comienzo del paciente con tirosinemia que nos fue trasladado desde otro centro.
ConclusionesLos pacientes con EMC pueden consultar en el SUP por diversos motivos, relacionados o no con su patología de base y que no siempre serán subsidiarios de un manejo especifico. Será de especial importancia intentar diferenciar en los casos de pacientes con EMC conocida si se trata de una descompensación aguda o bien de un proceso intercurrente que asocie riesgo de descompensación, siendo los síntomas en muchas ocasiones superponibles e inespecíficos. El comienzo de una EMC en el SUP es una situación extremadamente infrecuente; no obstante, se debe contar con protocolos de actuación ante el paciente con signos o síntomas indicativos. Las EMC son patologías complejas, cuyo manejo implica el conocimiento de las principales complicaciones metabólicas potencialmente graves o que puedan poner en riesgo la vida del paciente.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
