
Advances in the early diagnosis and treatment have led to improved survival, and a better quality of life for patients with inherited metabolic disorders (IMD). They can go to the Pediatric Emergency Services (PES) for reasons unrelated to their disease. The purpose of this study was to review the characteristics of visitors to the PES of these patients in a tertiary hospital.
Material and methodsA retrospective observational study was conducted on all visits from patients with IMD to the PES of Hospital Infantil La Paz over the years 2011 and 2012. IMD type, complaint, duration of symptoms, need for hospitalisation, and presence of metabolic decompensation were recorded.
ResultsA total of 107 visits were analysed, with the most frequent reason being for consultation of respiratory processes (30.8%). When the consultation was for vomiting, patients with protein-related disorders were those who delayed less in going to PES. One third of visitors were admitted, half of them due to metabolic decompensation of the underlying pathology.
ConclusionsPatients with IMD came to PES for many different reasons, which in some cases were the cause or consequence of an acute metabolic decompensation that led to hospitalisation. Being diseases with low prevalence, it would be useful to have diagnostic and therapeutic protocols in order to provide optimal care.
Los avances en el diagnóstico precoz y tratamiento han propiciado una mayor supervivencia, y en mejores condiciones, de los pacientes con enfermedades metabólicas congénitas (EMC). Estos pueden acudir a los Servicios de Urgencias Pediátricas (SUP) por motivos relacionados o no con su enfermedad. El propósito de este trabajo fue revisar las características de las visitas al SUP de estos pacientes, en un hospital de tercer nivel.
Material y métodosSe desarrolló un estudio observacional retrospectivo en el que se analizaron todas las visitas al SUP del Hospital Infantil La Paz durante los años 2011 y 2012 de pacientes con EMC. Se registraron el tipo de EMC, el motivo de consulta, el tiempo de evolución de los síntomas, la necesidad de ingreso hospitalario y la presencia de descompensación metabólica.
ResultadosEn total, fueron analizadas 107 visitas, siendo el motivo de consulta más frecuente los procesos respiratorios (30,8%). Cuando la consulta fue por vómitos, los pacientes con trastornos relacionados con las proteínas fueron los que menos tardaron en acudir al SUP. Un tercio de las visitas se siguió de ingreso, siendo la mitad de ellas por descompensación metabólica de la patología de base.
ConclusionesLos pacientes con EMC acudieron al SUP por motivos muy diversos, que en algunos casos fueron causa o consecuencia de una descompensación metabólica aguda que motivó el ingreso hospitalario. Al tratarse de enfermedades con baja prevalencia individual, resulta de interés contar con protocolos diagnóstico-terapéuticos que faciliten una atención óptima.
Inherited metabolic disorders (IMDs) are hereditary diseases with a genetic basis. Their individual prevalence is low, but combined they occur in approximately 1 in 600 live births.1,2 Until a few years ago, they often resulted in death or the development of severe sequelae and significant disability. At present, we have tools that increasingly allow for early diagnosis and initiation of treatments that improve quality of life and clinical outcomes in these patients. As a consequence, there is an increasing number of visits to paediatrics and paediatric emergency departments by children diagnosed with IMDs seeking care for various reasons that may or may not be related to their underlying disease.3 This increase has been intensified since the introduction of expanded neonatal screening programmes that have allowed for the survival of many patients with a good quality of life.4–6 It is essential that paediatric emergency care providers be familiar with this population, as these patients may require urgent care even when presenting with mild complaints, as these can cause metabolic decompensation in the context of their underlying disease.7
The aim of this study was to review the rate of Paediatric Emergency Department (PED) visits in children diagnosed with IMDs.
Patients and methodsWe conducted a retrospective observational study by reviewing the visits to the PED of the Hospital Universitario La Paz made in 2011 and 2012 by patients diagnosed with IMD that were being followed up in the hospital's Department of Childhood Nutrition and Metabolic Disorders during that period. Our study did not include patients with genetic dyslipidaemias.
During the period of the study, 58 patients with IMDs were followed up in the Nutrition and Metabolic Disorders Department. We classified these patients in four groups according to their underlying disease:
- 1.
Carbohydrate metabolism disorders (CMDs): patients with glycogen storage diseases, disorders of galactose metabolism, disorders of fructose metabolism, congenital hyperinsulinism and intracellular glucose transport disorders. Total: 7 patients.
- 2.
Lipid metabolism disorders (LMDs): patients with fatty acid beta-oxidation disorders, disorders of carnitine metabolism and disorders of ketone body synthesis and utilisation. Total: 8 patients.
- 3.
Protein/amino acid metabolism disorders (P/AAMDs): patients with hyperphenylalaninaemia, disorders of tyrosine metabolism, nonketotic hyperglycinaemia, propionic acidaemia, methylmalonic acidaemia, disorders of vitamin B12 intracellular metabolism and transport, maple syrup urine disease, methylcrotonylglycinuria, glutaric aciduria type 1, classical homocystinuria and disorders of sulphur-bearing amino acid metabolism, histidinaemia and urea cycle disorders. Total: 40 patients.
- 4.
Mitochondrial diseases (MDs): patients with isolated or combined respiratory chain and oxidative phosphorylation deficiencies, and patients with pyruvate dehydrogenase deficiency. Total: 3 patients.
In addition to these patients, the caseload of the department included five patients with disorders of complex molecule metabolism, specifically with congenital disorders of glycosylation and lysosomal storage disease. Of these patients, only one with a congenital disorder of glycosylation type IA visited the PED during the period under study. We included this patient in the P/AAMD group for the statistical analysis.
For each patient, we recorded the number of visits to the PED during the period under study, the reason for the visit, the duration of symptoms, admission to the hospital from the PED and the presence of associated metabolic decompensation. We performed the statistical analysis with the StatCalc 5.0 software. We used the chi-square test for the comparison of nonparametric variables. The level of statistical significance was set at P<.05.
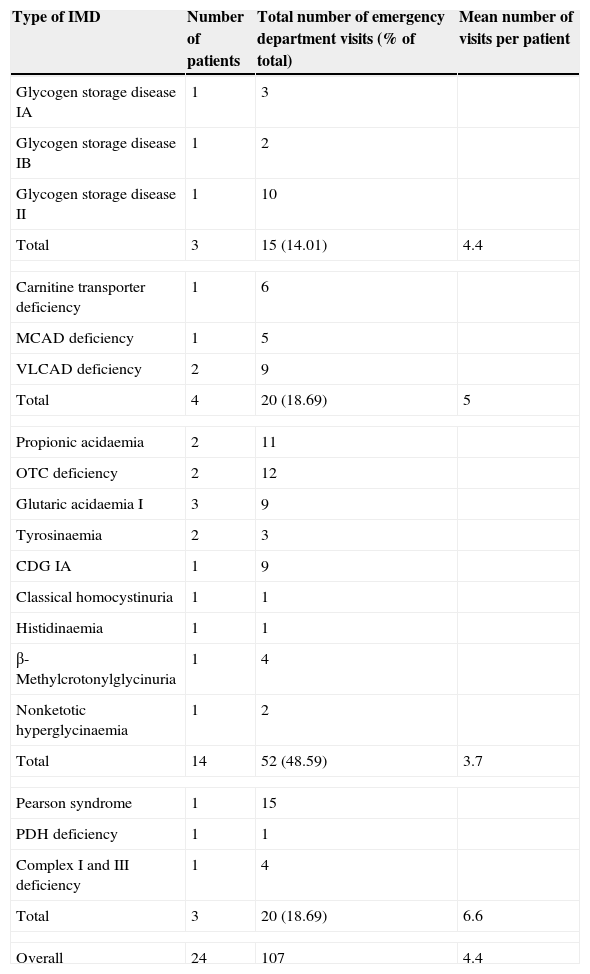
ResultsA total of 24 patients were included in our review (Table 1). The P/AAMD group was the largest (14 patients), with ages ranging from 2 months (infant admitted with liver failure that was later diagnosed with tyrosinaemia type 1) to 16 years and 10 months (age at last visit of a patient with ornithine transcarbamylase [OTC] deficiency). The mean age of patients in this group was 11.9 years.
Patients with IMDs that received care at the emergency department (2011–2012). Association between type of IMD and frequency of emergency department visits (n=107 visits).
| Type of IMD | Number of patients | Total number of emergency department visits (% of total) | Mean number of visits per patient |
|---|---|---|---|
| Glycogen storage disease IA | 1 | 3 | |
| Glycogen storage disease IB | 1 | 2 | |
| Glycogen storage disease II | 1 | 10 | |
| Total | 3 | 15 (14.01) | 4.4 |
| Carnitine transporter deficiency | 1 | 6 | |
| MCAD deficiency | 1 | 5 | |
| VLCAD deficiency | 2 | 9 | |
| Total | 4 | 20 (18.69) | 5 |
| Propionic acidaemia | 2 | 11 | |
| OTC deficiency | 2 | 12 | |
| Glutaric acidaemia I | 3 | 9 | |
| Tyrosinaemia | 2 | 3 | |
| CDG IA | 1 | 9 | |
| Classical homocystinuria | 1 | 1 | |
| Histidinaemia | 1 | 1 | |
| β-Methylcrotonylglycinuria | 1 | 4 | |
| Nonketotic hyperglycinaemia | 1 | 2 | |
| Total | 14 | 52 (48.59) | 3.7 |
| Pearson syndrome | 1 | 15 | |
| PDH deficiency | 1 | 1 | |
| Complex I and III deficiency | 1 | 4 | |
| Total | 3 | 20 (18.69) | 6.6 |
| Overall | 24 | 107 | 4.4 |
CDG IA, congenital disorder of glycosylation type IA; MCAD, medium-chain acyl-CoA dehydrogenase; OTC, ornithine transcarbamylase; PDH, pyruvate dehydrogenase; VLCAD, very long-chain acyl-coA dehydrogenase.
The LMD group, which comprised four patients, was the most homogeneous in age (mean, 1.2 years; median, 0.9 years; mode, 0.8 years).
The group of CMD included 3 patients: one 16-year-old male with glycogen storage disease type IA, a 3-year-old boy with glycogen storage disease type IB, and a girl with glycogen storage disease type II (who made the most visits to the PED in this group, 10 in total) that was 6 months old at the time of her first visit and 1 year and 7 months in her last visit.
The patient that made the most visits to the PED (15) belonged to the MD group, a boy with Pearson syndrome that made the first visit at 8 months and the last one at 2 years and 8 months of age. The mean age of patients in the MD group was 6 years.
Association between congenital metabolic disorder group and rate of emergency department visitsDuring the period under study, of the 58 patients with IMDs followed up at the Nutrition and Metabolic Disorders Department, 24 (41%) visited the PED for a total of 107 visits. Patients with P/AAMDs made the most visits overall, amounting to 48.5% of the total (52/107). However, when we analysed the number of visits per patient (vpp) we found that the highest mean number of visits corresponded to patients in the MD group (6.6 vpp) followed by patients in the LMD group (5 vpp), the CMD group (4.4 vpp) and lastly the P/AAMD group (3.7 vpp) (Table 1).
The visit rates were similar when we considered all the patients that were being followed up in the department during the period under study (MD, 6.6 vpp; LMD, 2.5 vpp; CMD, 2.1 vpp; P/AAMD, 1.3 vpp).
Visits were most frequent during the afternoon shift (39% of the visits), followed by the morning shift (33.6%) and least frequent during the night shift (27.1%). The mean time elapsed since the onset of symptoms of underlying IMDs was similar in the CMD, P/AAMD and MD groups (42, 32 and 29 months, respectively).
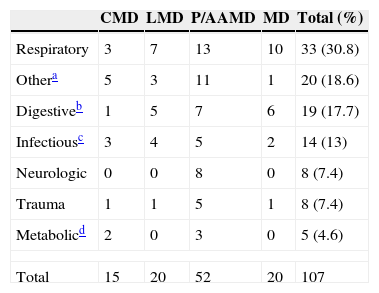
Analysis of reasons for PED visitsThe most common reasons for PED visits involved respiratory conditions (30.8%). This group included all visits due to respiratory symptoms with or without fever, refusal to feed and/or vomiting and respiratory distress (Table 2). Most of these patients did not require specific therapeutic interventions for their underlying disease and were managed similarly to patients without IMDs.
Reasons for PED visits in patients with IMDs (number of visits).
| CMD | LMD | P/AAMD | MD | Total (%) | |
|---|---|---|---|---|---|
| Respiratory | 3 | 7 | 13 | 10 | 33 (30.8) |
| Othera | 5 | 3 | 11 | 1 | 20 (18.6) |
| Digestiveb | 1 | 5 | 7 | 6 | 19 (17.7) |
| Infectiousc | 3 | 4 | 5 | 2 | 14 (13) |
| Neurologic | 0 | 0 | 8 | 0 | 8 (7.4) |
| Trauma | 1 | 1 | 5 | 1 | 8 (7.4) |
| Metabolicd | 2 | 0 | 3 | 0 | 5 (4.6) |
| Total | 15 | 20 | 52 | 20 | 107 |
CMD, carbohydrate metabolism disorder; LMD, lipid metabolism disorder; MD, mitochondrial disease; P/AAMD: protein/amino acid metabolism disorder.
This category included the following presenting complaints: crying/irritability, dermatologic symptoms, asthenia/weakness, nontraumatic musculoskeletal symptoms, feeding device problems, as well as other reasons for visiting the PED: treatment adjustments and a patient that sought care for a chance detection of a mass in the floor of the mouth.
Digestive symptoms comprised the second most frequent group of reasons for PED visits. Among them, isolated vomiting was the leading complaint (11.21%), and we found that the time elapsed from onset of these symptoms was lower in patients with P/AAMD (mean time since onset of vomiting, 8.4h) compared to patients from the other three groups (CMD, 60h; LMD, 48h; MD, 69.6h).
Other reasons for PED visitsIn addition to visits related to traumatic injury (8 visits), there was a broad range of other presenting complaints (crying/irritability, dermatologic symptoms, asthenia/general malaise, musculoskeletal problems of nontraumatic aetiology, urinary symptoms, questions or concerns regarding treatment, etc.) that accounted for a total of 20 visits. Among them, the most frequent reasons involved problems with feeding devices (9/20; 45%).
None of the patients seeking care for a musculoskeletal, dermatologic or urinary problem required specific interventions due to their underlying metabolic disease.
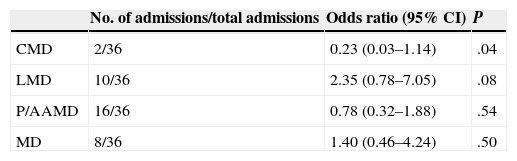
Admission of patients with inherited metabolic disorders from the Paediatric Emergency DepartmentHospital admission resulted from 33.64% (36/107) of the visits to the PED. In our analysis of the association between the type of IMD and the risk of admission, the association was statistically significant for the CMD group (Table 3).
Risk of admission by type of IMD.
| No. of admissions/total admissions | Odds ratio (95% CI) | P | |
|---|---|---|---|
| CMD | 2/36 | 0.23 (0.03–1.14) | .04 |
| LMD | 10/36 | 2.35 (0.78–7.05) | .08 |
| P/AAMD | 16/36 | 0.78 (0.32–1.88) | .54 |
| MD | 8/36 | 1.40 (0.46–4.24) | .50 |
CMD, carbohydrate metabolism disorder; LMD, lipid metabolism disorder; MD, mitochondrial disease; P/AAMD: protein/amino acid metabolism disorder.
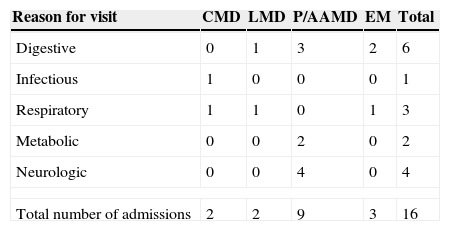
Of the 36 admissions, 16 (44.4%) were due to metabolic decompensation of the underlying disease (Table 4). The reason for PED visits associated most frequently with decompensation was the presence of digestive symptoms (6/16 visits; 37.5%), and particularly isolated vomiting, followed by neurologic (4/16 visits; 25%) and respiratory symptoms (3/16 visits; 18.7%). The mean time elapsed from the onset of symptoms to the PED visit in patients admitted for metabolic decompensation was 27.13h.
Total number of admissions due to metabolic decompensation by initial reason for emergency department visit.
| Reason for visit | CMD | LMD | P/AAMD | EM | Total |
|---|---|---|---|---|---|
| Digestive | 0 | 1 | 3 | 2 | 6 |
| Infectious | 1 | 0 | 0 | 0 | 1 |
| Respiratory | 1 | 1 | 0 | 1 | 3 |
| Metabolic | 0 | 0 | 2 | 0 | 2 |
| Neurologic | 0 | 0 | 4 | 0 | 4 |
| Total number of admissions | 2 | 2 | 9 | 3 | 16 |
CMD, carbohydrate metabolism disorder; LMD, lipid metabolism disorder; MD, mitochondrial disease; P/AAMD: protein/amino acid metabolism disorder.
The characteristics of patients admitted with metabolic decompensation differed based on their underlying disease (Table 5):
- 1.
CMD: 2 admissions. The first one was a patient with glycogen storage disease type IA that sought care for fever of recent onset accompanied by respiratory symptoms and vomiting, in the context of poor glycaemic control in the home in the days preceding the visit. The emergency department findings included normal glucose and metabolic acidosis. The second patient had glycogen storage disease type IB and also sought care for febrile disease with vomiting; his parents had not detected low glucose levels at home. PED findings included hypoglycaemia (54mg/dL) accompanied by hyperlactacidaemia and compensated metabolic acidosis. Fluid therapy was initiated, and poor oral tolerance prompted admission of the patient.
- 2.
LMD: 2 admissions. The first one involved a newborn 28 days of age with very long-chain acyl-coA dehydrogenase (VLCAD) deficiency that had sought care for fever and respiratory symptoms accompanied by feeding difficulties. Her initial glucose level was 54mg/dL, and intravenous fluid therapy with dextrose supplementation (7mg/kg/min) was initiated. Arterial blood gas and lactate measurements were normal. The second patient had carnitine transporter deficiency and sought care for isolated vomiting. The PED findings included hyperlactacidaemia, acidosis, and normal glucose.
- 3.
P/AAMD: 9 admissions, 8 of which were due to hyperammonaemia (normal range for age >1 month in our laboratory, 15–45μg/dL) in 2 patients with ornithine transcarbamylase deficiency and 1 patient with propionic acidaemia (none of whom were younger than 1 month). The other admission involved an infant 2 months of age that was transferred to our hospital for a coagulation study. The original reason for care seeking had been an episode of potential choking associated with an absence seizure and hypotonia, which prompted the initial laboratory tests. Once in the PED, the neurologic findings were normal, and a hepatomegaly of 1–2cm was detected. A second round of laboratory tests confirmed the presence of clotting abnormalities and the patient was admitted for a coagulation study. The investigation confirmed the diagnosis of tyrosinaemia type 1.
- 4.
MD: 3 admissions, all corresponding to a single patient with Pearson syndrome. The first time, the reason for the visit had been isolated vomiting, and hyponatraemia (128mmol/L) associated to lactic acid with compensatory respiratory alkalosis which was detected in the PED. In the second admission, the decompensation was caused by metabolic acidosis in the context of a respiratory infection with associated vomiting. The third admission was due to a severe episode of lactic acidosis associated with severe hypoglycaemia (11mg/dL) secondary to gastroenteritis.
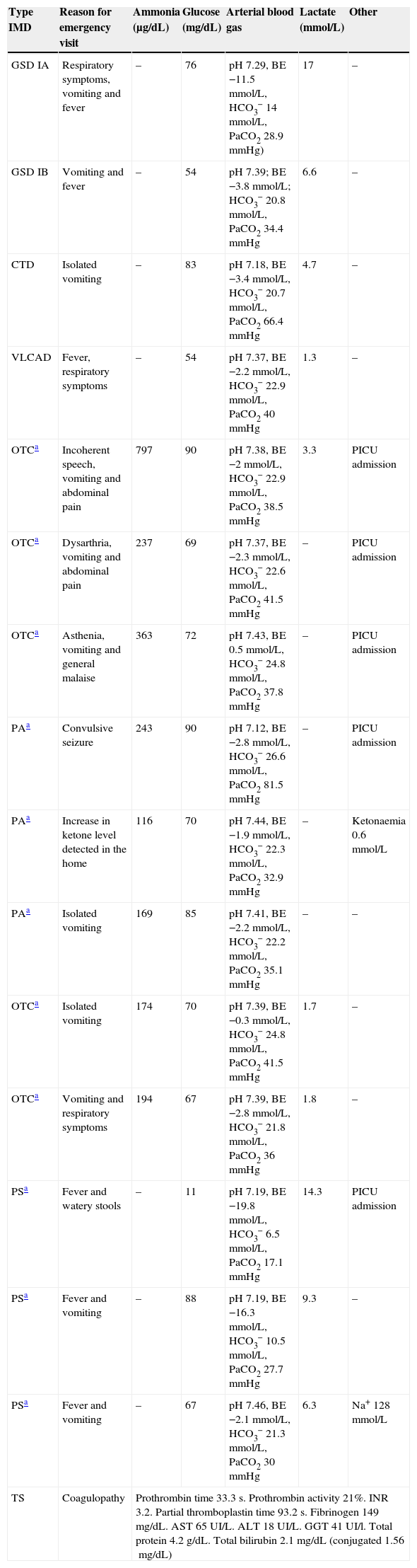
Admissions for metabolic decompensation of patients with IMD seen at the emergency department (2011–2012). Description of decompensation.
| Type IMD | Reason for emergency visit | Ammonia (μg/dL) | Glucose (mg/dL) | Arterial blood gas | Lactate (mmol/L) | Other |
|---|---|---|---|---|---|---|
| GSD IA | Respiratory symptoms, vomiting and fever | – | 76 | pH 7.29, BE −11.5mmol/L, HCO3− 14mmol/L, PaCO2 28.9mmHg) | 17 | – |
| GSD IB | Vomiting and fever | – | 54 | pH 7.39; BE −3.8mmol/L; HCO3− 20.8mmol/L, PaCO2 34.4mmHg | 6.6 | – |
| CTD | Isolated vomiting | – | 83 | pH 7.18, BE −3.4mmol/L, HCO3− 20.7mmol/L, PaCO2 66.4mmHg | 4.7 | – |
| VLCAD | Fever, respiratory symptoms | – | 54 | pH 7.37, BE −2.2mmol/L, HCO3− 22.9mmol/L, PaCO2 40mmHg | 1.3 | – |
| OTCa | Incoherent speech, vomiting and abdominal pain | 797 | 90 | pH 7.38, BE −2mmol/L, HCO3− 22.9mmol/L, PaCO2 38.5mmHg | 3.3 | PICU admission |
| OTCa | Dysarthria, vomiting and abdominal pain | 237 | 69 | pH 7.37, BE −2.3mmol/L, HCO3− 22.6mmol/L, PaCO2 41.5mmHg | – | PICU admission |
| OTCa | Asthenia, vomiting and general malaise | 363 | 72 | pH 7.43, BE 0.5mmol/L, HCO3− 24.8mmol/L, PaCO2 37.8mmHg | – | PICU admission |
| PAa | Convulsive seizure | 243 | 90 | pH 7.12, BE −2.8mmol/L, HCO3− 26.6mmol/L, PaCO2 81.5mmHg | – | PICU admission |
| PAa | Increase in ketone level detected in the home | 116 | 70 | pH 7.44, BE −1.9mmol/L, HCO3− 22.3mmol/L, PaCO2 32.9mmHg | – | Ketonaemia 0.6mmol/L |
| PAa | Isolated vomiting | 169 | 85 | pH 7.41, BE −2.2mmol/L, HCO3− 22.2mmol/L, PaCO2 35.1mmHg | – | – |
| OTCa | Isolated vomiting | 174 | 70 | pH 7.39, BE −0.3mmol/L, HCO3− 24.8mmol/L, PaCO2 41.5mmHg | 1.7 | – |
| OTCa | Vomiting and respiratory symptoms | 194 | 67 | pH 7.39, BE −2.8mmol/L, HCO3− 21.8mmol/L, PaCO2 36mmHg | 1.8 | – |
| PSa | Fever and watery stools | – | 11 | pH 7.19, BE −19.8mmol/L, HCO3− 6.5mmol/L, PaCO2 17.1mmHg | 14.3 | PICU admission |
| PSa | Fever and vomiting | – | 88 | pH 7.19, BE −16.3mmol/L, HCO3− 10.5mmol/L, PaCO2 27.7mmHg | 9.3 | – |
| PSa | Fever and vomiting | – | 67 | pH 7.46, BE −2.1mmol/L, HCO3− 21.3mmol/L, PaCO2 30mmHg | 6.3 | Na+ 128mmol/L |
| TS | Coagulopathy | Prothrombin time 33.3s. Prothrombin activity 21%. INR 3.2. Partial thromboplastin time 93.2s. Fibrinogen 149mg/dL. AST 65UI/L. ALT 18UI/L. GGT 41UI/l. Total protein 4.2g/dL. Total bilirubin 2.1mg/dL (conjugated 1.56mg/dL) | ||||
ALT, alanine aminotransferase; AST, aspartate aminotransferase; BE, base excess; CTD, carnitine transport deficiency; MD, metabolic decompensation; GSD IA, glycogen storage disease type IA; GSD IB, glycogen storage disease type IB; GGT, gamma-glutamyl transpeptidase; INR, international normalised ratio; OTC, ornithine transcarbamylase deficiency; PA, propionic acidaemia; PICU, paediatric intensive care unit; PS, Pearson syndrome; TS, tyrosinaemia type I; VLCAD, very long-chain acyl-coA dehydrogenase deficiency.
In the subset of admissions that were not due to metabolic decompensation (20/36; 55.5%), 60% (12/20) were due to fever and gastrointestinal conditions, corresponding to visits by 4 patients with MD and 4 patients with LMD. In 41.6% of these cases (5/12) the reason for admission was the risk of metabolic decompensation due to poor oral tolerance. Respiratory conditions accounted for 25% (5/20) of admissions in this subset, none of which were related to a risk of decompensation: 2 patients (MCAD deficiency and Pearson syndrome) with pneumonia that required intravenous antibiotic treatment, and 3 patients (propionic acidaemia, classical homocystinuria, and glutaric acidaemia) with acute respiratory failure that required oxygen therapy.
DiscussionWe did not find statistically significant differences associating the type of IMD with a greater risk of admission. While the association was statistically significant for the CMD group, this result was of little clinical relevance because the odds ratio (for an observational study) was very small, and the confidence interval was virtually nonexistent.
Patients with IMDs tend to visit the PED for the slightest acute alterations in general health status due to the possibility of them leading to metabolic decompensation.8 In this context, emergency management differs considerably from that of a child without IMD, as the goal of the paediatrician is the early prevention or detection of a potential decompensation and the treatment of any alterations that may threaten the life of the patient.9–11 One or more of the following biochemical abnormalities were detected in most of the patients in this series: acid–base imbalance or coagulopathy, hyperammonaemia, hypoglycaemia, and electrolyte abnormalities.12,13 It is sometimes difficult to determine whether the presenting symptoms of the patient are secondary to decompensation or to the associated intercurrent condition.
Hyperammonaemia was observed in patients in the P/AAMD group, specifically those with urea cycle disorders14–17 and organic acidaemias.17,18 Patients with urea cycle disorders sought care for neurologic symptoms or isolated vomiting (both manifestations can be symptomatic of elevated blood ammonia levels), while the decision to visit the PED in patients with organic acidaemias was due to increased ketonaemia, irrespective of the intensity of the accompanying symptoms (mostly cold symptoms and isolated vomiting). Hyperammonaemia may be accompanied by respiratory alkalosis due to the stimulating effect of ammonia on the brainstem respiratory centres, and thus the presence of tachypnoea in a patient with IMD should not be underestimated.19
Hypoglycaemia can be found in different IMDs and delays in emergency treatment can result in permanent brain damage.20,21 In our series, there were two admissions due to hypoglycaemia in patients from the CMD group, both of whom had glycogen storage disease type I. In this disease, defects in the glucose-6-phosphatase complex result in the inability to produce glucose from glycogenolysis and gluconeogenesis, which activates lipid catabolism pathways. Biochemical findings include metabolic acidosis, hyperlactacidaemia and dyslipidaemia. Patients with glycogen storage disease type IB may also develop neutropaenia, a predisposing factor for severe bacterial infections; in our series, the neutrophil count of the patient with glycogen storage disease type IB was normal (1510/μL, with a total white blood cell count of 5000/μL).
The remaining episodes of hypoglycaemia were observed in one patient with Pearson syndrome and one patient with VLCAD deficiency. Pearson syndrome is a rare multiorgan mitochondrial cytopathy in which hypoglycaemic events are associated with liver involvement. In VLCAD deficiency, episodes of hypoketotic hypoglycaemia22 may be triggered by fasting, prolonged physical exertion or intercurrent disease. Patients with disorders of carnitine metabolism are also at risk of developing hypoketotic hypoglycaemia secondary to defects in the beta-oxidation of long-chain fatty acids. Thus, the prevention of hypoglycaemia by having frequent meals rich in complex carbohydrates and avoiding long periods of fasting is recommended in the management of both diseases.
Metabolic acidosis is a common finding in ill children, and it is essential that the presence or absence of ketones and an anion gap is determined in the course of its investigation. Metabolic acidosis with a normal anion gap should be interpreted as an indicator of bicarbonate loss through the kidney or the gastrointestinal tract.9,23 However, a high anion gap is usually present in IMDs that indicates the presence of abnormal metabolites like ketoacids, lactic acid or the organic acid that cannot be metabolised.8 Metabolic acidosis may occur in disorders of amino acid or pyruvate metabolism, mitochondrial diseases and also disorders of carbohydrate metabolism.17,24,25 Lactic acidosis is a frequent finding in mitochondrial diseases, glycogen storage disorders and gluconeogenesis disorders,24,25 among others.
During the period under study, no patient was newly diagnosed with IMD during their first visit to our PED, except for the onset of tyrosinaemia in one patient that was transferred from another facility.
ConclusionsPatients with IMDs visit PEDs for various reasons that may or may not be related with their underlying disease and that do not always require specific management. In patients with a known IMD, it is particularly important to differentiate between acute decompensation and intercurrent diseases that carry a risk of decompensation, as the symptoms often overlap and are nonspecific. It is very rare for IMDs to be first identified at a PED; nevertheless, protocols must be in place for the management of patients that present with signs or symptoms suggestive of IMD. Inherited metabolic disorders are complex diseases, and their management requires knowledge of the most common metabolic complications that can be severe or life-threatening.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Molina Gutiérrez MA, López López R, Morais López A, Bueno Barriocanal M, Martínez Ojinaga Nodal E, Alcolea Sánchez AM, et al. Enfermedades metabólicas congénitas en un servicio de urgencias pediátricas. An Pediatr (Barc). 2015;82:404–411.
