



La enfermedad de Niemann-Pick tipo C está causada por un defecto en el transporte intracelular de colesterol que produce un acúmulo de lípidos en los lisosomas de diferentes tejidos. Es una enfermedad rara, debida generalmente a mutaciones en el gen NPC1 y solo unos pocos casos se asocian a mutaciones en el gen NPC2. Frecuentemente se manifiesta en la edad pediátrica, presentando gran variabilidad en las manifestaciones clínicas. La enfermedad conduce a un deterioro neurológico con diferentes síntomas que están relacionados con la edad. Una colestasis neonatal transitoria, la aparición de esplenomegalia y/o hepatomegalia pueden preceder en años a los síntomas neurológicos.
Pacientes y métodosPresentamos los 6 casos diagnosticados en nuestra unidad en los últimos 20 años. Se han revisado las manifestaciones clínicas, los hallazgos neurorradiológicos (RM) y el análisis molecular de todos ellos.
ResultadosTodos se presentaron antes de los 6 años y 5 casos tuvieron afectación hepática y/o colestasis en el periodo neonatal. En 2 casos se detectó ascitis en el periodo prenatal. La presencia de esplenomegalia se objetivó en 5 casos. En todos los casos se detectaron mutaciones en el gen NPC1.
ConclusiónEs importante el conocimiento de esta enfermedad y la identificación de los síntomas clínicos precoces para poder diagnosticarla precozmente, lo que conllevaría a un tratamiento adecuado, pudiendo evitar procedimientos innecesarios. Por otra parte es importante asesorar adecuadamente a las familias y proporcionar un consejo genético.
Niemann-Pick type C is a lysosomal storage disorder caused by a defect in intracellular trafficking of cholesterol. It is a rare disease, usually caused by mutations in NPC1 gene, but in some cases by mutations in NPC2 gene. Usually it is present in the paediatric age with a great variability of clinical manifestations. This disease leads to neurological degeneration with various age-related symptoms. Transient neonatal cholestasis, the appearance of splenomegaly and/or hepatomegaly may occur years before the neurological symptoms.
Patients and methodsWe report 6 cases diagnosed in our unit in the last 20 years. We reviewed the clinical manifestations, neuroradiological findings (MRI) and molecular analysis of all of them.
ResultsThe disease began before 6 years of age and 5 cases had liver dysfunction and cholestasis in the neonatal period. Ascites was detected in 2 cases in prenatal period. Five cases have or had splenomegaly. Mutations in NPC1 gene were detected in all of them.
ConclusionsIt is important to understand this disease and the identification of early clinical symptoms to make an early diagnosis, leading to appropriate treatment and avoiding unnecessary tests. Moreover, it is important to suitably advise families and provide them with genetic counselling.
La enfermedad de Niemann-Pick tipo C (NPC) es debida a un defecto en el trasporte intracelular de colesterol, esfingolípidos y glicoesfingolípidos, que se acumulan junto al colesterol no esterificado en los endosomas tardíos y lisosomas1. La incidencia estimada de la enfermedad es de 1/150.000 recién nacidos vivos en Europa. Se hereda de forma autosómica recesiva y es producida en la mayoría de los casos (95%) por mutaciones en el gen NPC1 y en una pequeña proporción (5%) en el NPC2. Afecta al sistema nervioso central, produciendo un deterioro neurológico y a otros órganos como el hígado y el bazo2,3.
Existe una gran variabilidad de manifestaciones clínicas4. La forma más frecuente de presentación asocia una colestasis neonatal transitoria, la aparición de esplenomegalia y/o hepatomegalia progresiva y posteriormente síntomas neurológicos. Entre las manifestaciones neurológicas destacan las alteraciones en la motilidad ocular siendo característica la parálisis de la mirada vertical, también pueden tener disfagia, disartria, ataxia, distonía, crisis epilépticas, daño cognitivo progresivo, síntomas psiquiátricos y crisis de cataplejía gelástica. La hepatoesplenomegalia es un signo frecuente, pero su ausencia no excluye el diagnóstico.
Se han establecido 5 formas clínicas5 dependiendo de la edad de presentación de los síntomas neurológicos. Forma infantil precoz (<2 años), forma infantil tardía (2–6 años), forma juvenil (6–16 años), forma adulta (>16 años). Además existe una forma perinatal de aparición precoz y con mala evolución clínica. En la tabla 1 se muestran los síntomas neurológicos y sistémicos más frecuentes en las distintas formas clínicas. A continuación presentamos los casos con enfermedad de NPC de nuestro centro con el objetivo de dar a conocer esta patología y alertar a los pediatras para realizar un diagnóstico precoz de la enfermedad.
Síntomas y signos clínicos de la enfermedad de NPC según las formas clínicas modificado de NP-C Guidelines Working Group5
| Formas clínicas | Manifestaciones sistémicas | Manifestaciones neurológicas |
| Pre/perinatal (≤3 meses) | Hidrops, hepatoesplenomegalia, ascitis fetal persistente o no al nacimiento, colestasis prolongada, insuficiencia respiratoria, fallo hepático | No reconocidas |
| Infantil precoz (3 meses a 2 años) | Hepatoesplenomegalia o hepatomegalia o esplenomegalia aislada | Retraso psicomotor, hipotonía central, hipoacusia, parálisis de la mirada vertical (rara) |
| Infantil tardío (2 años a 6 años) | Visceromegalia (frecuente) | Caídas frecuentes, torpeza motora, ataxia, distonía, disfagia y disartria progresivas, hipotonía central, hipoacusia, crisis convulsivas, cataplejía, parálisis de la mirada vertical |
| Juvenil (6 a 15 años) | Visceromegalia (no siempre) | Fracaso escolar, problemas de aprendizaje, problemas de conducta, caídas frecuentes, torpeza motora, ataxia, disartria, distonía, disfagia, mioclonías, cataplejía, crisis convulsivas, parálisis de la mirada vertical |
| Adolescente y adultos (>15 años) | Visceromegalia (no siempre presente) o esplenomegalia aislada (en algunos casos) | Torpeza, cataplejía, síntomas psiquiátricos, deterioro cognitivo, demencia, parálisis de la mirada vertical, ataxia, distonía, disartria, disfagia, mioclonías, crisis (parciales o generalizadas) |
Desde el año 1990 se han diagnosticado 6 pacientes (de entre 0 y 12 años de edad) con enfermedad de NPC en nuestra unidad. Hemos realizado un estudio descriptivo mediante la revisión de las historias clínicas. En todos los casos se ha confirmado el diagnóstico en fibroblastos cultivados demostrando un acúmulo de colesterol no esterificado en los lisosomas después de realizar el análisis ciotoquímico con filipina6. Mediante la utilización del microscopio óptico con luz ultravioleta se puede observar un acúmulo de vesículas perinucleares fluorescente que indican la localización y el aumento cualitativo de colesterol no esterificado. Se define «fenotipo clásico» a los casos que presenta la tinción con filipina claramente positiva y «fenotipo variante» a los casos en los que el aumento de vesículas fluorescente es muy escaso o prácticamente nulo. El diagnóstico bioquímico se confirmó mediante el análisis molecular. El diagnóstico molecular se realizó a través de la amplificación del DNA a través de la reacción en cadena de la polimerasa (PCR) de los distintos exones y zonas intrónicas flanqueantes del gen NPC1 y posterior secuenciación de dichos fragmentos siguiendo protocolos previamente establecidos7. Se han revisado las manifestaciones clínicas, los hallazgos neurorradiológicos (RM) y el análisis molecular de todos ellos. Se ha solicitado consentimiento informado a los familiares de acuerdo con el comité de ética del centro.
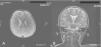
ResultadosCaso 1: niño de 3 años de edad que presentó ascitis masiva y hepatoesplenomegalia sin otros signos de hidrops fetal en la semana 20 de gestación. El parto fue en la semana 31 mediante cesárea. Durante el periodo neonatal presentó ascitis de difícil control, colestasis neonatal y disfunción hepática. Se le realizó biopsia hepática que mostró intensa colestasis hepatocitaria y canalicular y leve ductopenia, sugestiva de pobreza de ductos biliares. Además presentó una hemorragia intraventricular grado ii/iii (debido a la prematuridad y a una coagulopatia secundaria a la afectación hepática). Durante el primer año de vida continuó con hepatoesplenomegalia, desarrollando una enfermedad crónica hepática y cirrosis con hipertensión portal además de retraso psicomotor severo. A los 2 años y medio sufrió un deterioro neurológico. En la exploración actual presenta hepatomegalia y esplenomegalia, espasticidad en miembros inferiores, temblor, distonía leve, sedestación inestable, estrabismo alternante, paresia de la mirada vertical y lesiones de dermatitis palpebral. Recientemente refiere crisis de cataplejía. Se realizó una RM cerebral a los 2,2 años donde se objetivó una alteración en la mielinización periventricular.
Caso 2: niña en la que se detectó ascitis masiva a las 30 semanas de gestación. Desde el nacimiento presentó gran hepatoesplenomegalia con hipertensión portal, instaurándose un fallo hepático que motivó 2 trasplantes hepáticos. Tras fallo del injerto falleció al 1,5 meses de vida. Los hallazgos anatomopatológicos en la biopsia hepática mostraron abundantes mielinosomas en el citoplasma de las células de Kupffer y en los hepatocitos correspondientes a lípidos parcialmente metabolizados en los lisosomas. Tras la autopsia se objetivó mediante microscopio electrónico abundante material de depósito en el citoplasma de las neuronas constituido por pequeños cuerpos pleomórficos (la mayor parte formados por membranas que se disponen concéntricamente).
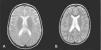
Caso 3: niña de 4 años. Durante el embarazo se objetivó un retraso del crecimiento intrauterino en el último trimestre. En el periodo neonatal presentó ictericia prolongada que no requirió ingreso. Desde los primeros meses de vida se objetivó escasa ganancia ponderal. Hacia los 3 meses se evidenció una gran hepatoesplenomegalia. A los 2 años se le realizó una esplenectomía, lo que precipitó una regresión neurológica importante. Ha tenido varios episodios de neumonía con insuficiencia respiratoria. En la actualidad presenta una encefalopatía con tetraparesia espástica, paresia de la mirada vertical y ausencia de lenguaje. Tiene crisis de cataplejía gelástica y crisis convulsivas. Se ha realizado RM cerebral a los 4 años en la que se ha objetivado atrofia corticosubcortical, fundamentalmente supratentorial, alteración en la mielinización que afecta predominantemente a la sustancia blanca posterior y subcortical. En la espectroscopia, hay descenso del pico de N-acetil-aspartato (Naa) tanto en ganglios basales como en centro semioval.
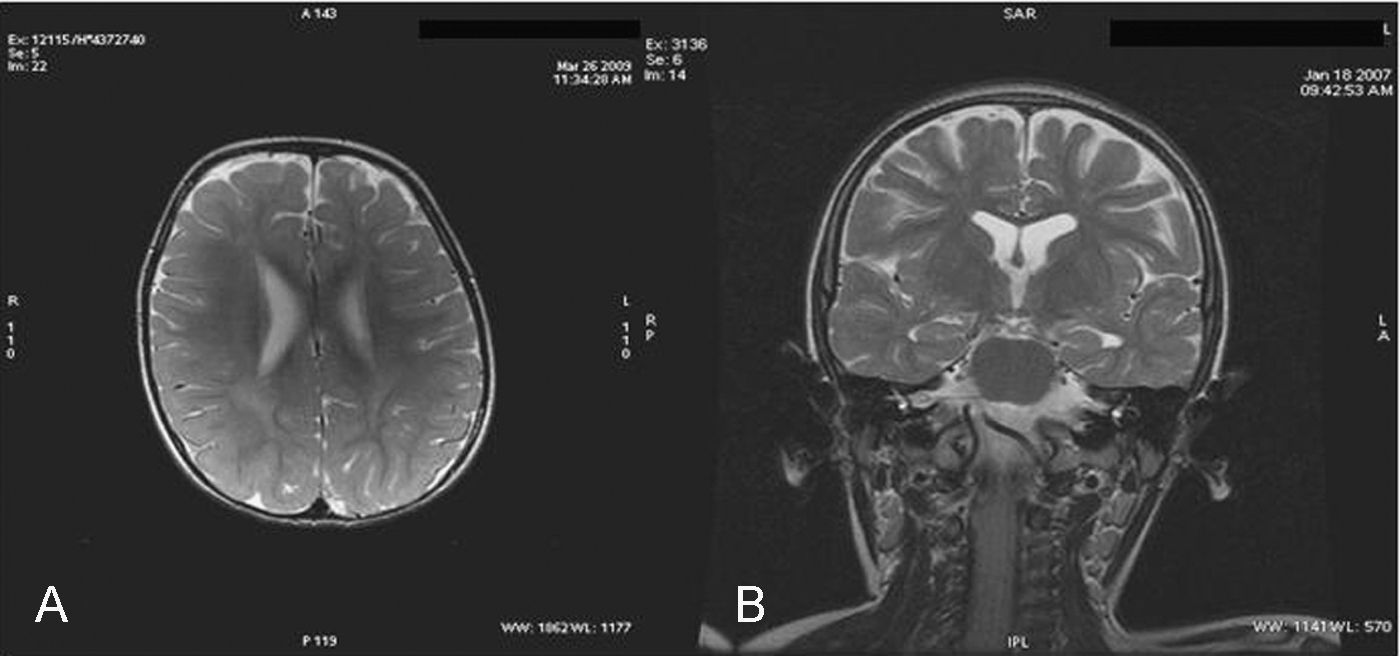
Caso 4: niño de 8 años. Desde el nacimiento presentó alteraciones en la pigmentación cutánea de forma lineal en el tronco (hemitórax izquierdo) y en la cara interna de la pierna sugerentes de hipomelanosis de ito. Durante el primer año de vida no le notaron nada anormal salvo estridor laríngeo ocasional. Desde el segundo año de vida se apreció un retraso del lenguaje y progresivamente torpeza motora. A partir de los 3 años de edad sufrió una regresión neurológica y ataxia. A los 5 años comenzó con alteración en la deglución y posteriormente con epilepsia mioclónica resistente a múltiples fármacos antiepilépticos. A la edad de 5 años acude por primera vez a nuestra consulta con el diagnóstico de hipomelanosis de ito y en la exploración presentaba esplenomegalia, déficit intelectual, ataxia, coordinación deficiente y ptosis palpebral leve del ojo derecho. A los 6 años desarrolló parálisis de la mirada vertical. En la RM cerebral realizada a los 5 años se objetivó atrofia mesial con dilatación del asta temporal.
Caso 5: niña de 5 años, de origen ecuatoriano, hija de padres consanguíneos. Desde el primer mes de vida presentó colestasis que remitió al año de vida, hepatoesplenomegalia y posteriormente retraso psicomotor severo con marcada hipotonía y ataxia. Le realizaron una biopsia hepática donde se objetivó moderada fibrosis portal y una intensa colestasis intrahepática con células espumosas sugestivas de enfermedad de depósito lipídico. A los 4 años comenzó con crisis de cataplejía gelástica, crisis epilépticas y parálisis de la mirada vertical. En la actualidad presenta hipotonía, ataxia y persiste la esplenomegalia. En la RM cerebral realizada a los 5 años se objetivó alteración severa de la mielinización.
Caso 6: niño de 12 años de edad. En el periodo neonatal presentó colestasis neonatal y posteriormente hepatopatía crónica con hepatoesplenomegalia. Se le realizó una biopsia hepática donde se objetivó una hepatopatía colestática con ductopenia. A los 5 años y medio comenzó con parálisis de la mirada vertical. A los 8 años inició una regresión neurológica con mal rendimiento escolar y déficit de atención seguido de deterioro cognitivo siendo remitido a nuestra consulta a los 9 años. A los 11 años presentó crisis convulsivas. En la exploración presenta ataxia, parálisis de la mirada vertical sin hepatoesplenomegalia. Se le ha realizado RM cerebral a los 9 años donde no se objetivaron alteraciones, salvo un quiste aracnoideo.
En todos los casos se observaron depósitos de colesterol no esterificado en fibroblastos mediante la tinción histoquímica con filipina, presentando todos un «fenotipo clásico» excepto el caso 4 que resultó ser un «fenotipo variante». El diagnóstico se confirmó con el estudio genético, hallándose mutaciones en el gen NPC1 en los 6 pacientes.
Todos los casos reciben actualmente tratamiento con miglustat (un ihibidor de la glucosilceramida sintasa responsable del primer paso de la síntesis de los glucolípidos). En la tabla 2 se muestra la edad de inicio de los síntomas extraneurológicos y neurológicos, edad de confirmación bioquímica, fenotipo bioquímico, análisis molecular e inicio de tratamiento de todos los casos.
Se muestran los síntomas extraneurológicos y neurológicos, edad de confirmación bioquímica, fenotipo bioquímico y análisis molecular
| Caso | 1 | 2 | 3 | 4 | 5 | 6 |
| Inicio síntomas EN | Prenatal | Prenatal | Prenatal | 2 años | Neonatal | Neonatal |
| 1.ersíntoma EN | ||||||
| Colestasis neonatal | + | + | + | + | + | |
| Ascitis fetal | + | + | ||||
| HE | + | + | ||||
| CIR | + | |||||
| Alt pigmentación | + | |||||
| Inicio síntomas N | 1 año | – | 2 años | 1 año | 1 año | 5,5 años |
| Síntomas N | ||||||
| Retraso psicomotor | + | + | + | |||
| Regresión | + | + | + | + | + | |
| Ataxia | + | + | + | |||
| Temblor | + | +/− | ||||
| Distonía | + | + | ||||
| Estrabismo | + | |||||
| PMV | + | + | + | + | + | |
| Tetraparesia | + | |||||
| Cataplejia | + | + | + | |||
| Crisis convulsivas | + | + | + | |||
| Edad diagnóstico | 3 meses | 3 meses | 2 años | 5,5 años | 1 año | 10 años |
| Fenotipo bioquímico22 | Clásico | Clásico | Clásico | Variante | Clásico | Clásico |
| Análisis molecular27 | p.[Q119VfsX8]+[D944N] | p.[G993EfsX4]+[G993EfsX4] | p.[Q775P]+[D944N] | p.[Q119VfsX8]+[G992W] | p.[C177Y]+[C177Y] | p.[N916del]+[A1151T] |
| Edad de inicio de tratamiento | 15 meses | – | 3 años | 6 años | 5 años | 10,5 años |
Alt: alteración; EN: extraneurológicos; HE: hepatoesplenomegalia; N: neurológicos; PMV: parálisis mirada vertical.
La enfermedad de NPC presenta una gran variedad de manifestaciones clínicas. Los síntomas extraneurológicos comenzaron en el periodo perinatal en 5 de los casos presentados. Todos menos el caso 4 presentaron colestasis neonatal y hepatopatía. Estos síntomas ayudan a un diagnóstico precoz de la enfermedad. En ocasiones es difícil discriminar cuáles son los primeros síntomas neurológicos debidos a la enfermedad de NPC en niños con retraso psicomotor previo, como en el caso 1. En el caso 2, con una forma perinatal y cuadro clínico muy grave, no se detectaron síntomas neurológicos, pero hay que destacar que aún en ausencia de los mismos en el examen neuropatológico se evidenciaron alteraciones propias de enfermedad de depósito en las neuronas.
Es importante remarcar que nuestra casuística apoya la experiencia de otros centros, en estos aconsejan que se debe pensar en esta enfermedad en cuadros de colestasis neonatal de etiología incierta8 y en las hepatitis idiopáticas neonatales9. En un estudio realizado en el Reino Unido objetivaron que la enfermedad de NPC es la segunda causa genética de enfermedad hepática en la infancia después del déficit de alfa-1 antitripsina10. Yerushalmi B refiere que la enfermedad de NPC es la causa más común de enfermedad metabólica/genética que se presenta como colestasis neonatal. Por otro lado el grado de afectación hepática no es un indicador de la gravedad de la progresión neurológica de la enfermedad9,11 (puesto que algunos evolucionan como formas adultas).
La esplenomegalia es un signo más consistente que la hepatomegalia en los niños, debemos tenerlo en cuenta para llegar a un diagnóstico precoz de la enfermedad12. Todos los casos vistos en nuestra unidad han tenido o tienen esplenomegalia. De hecho el caso 3 sufrió una regresión neurológica rápida precipitada tras la esplenectomía por el acúmulo de lípidos en el sistema nervioso.
Entre los síntomas neurológicos la parálisis de la mirada vertical es un signo característico que suele aparecer de 2 a 4 años después de los signos de afectación cerebelosa y muy próximo al inicio de otros síntomas secundarios a afectación de tronco cerebral13. Se ha objetivado en todos nuestros casos excepto en el caso 2. Las crisis de cataplejía gelástica aparecieron en el caso 3 a la edad de 3 años y en el caso 5 a la edad de 4 años, similar a lo referido en otros artículos13.
La progresión de la enfermedad es más rápida cuando los síntomas se inician precozmente14. El único tratamiento que existe actualmente es la terapia de reducción de sustrato con miglustat15,16. Este fármaco está aprobado como medicamento de uso compasivo.
En un estudio reciente se ha demostrado enlentecimiento en la manifestación de los síntomas neurológicos de la enfermedad17. Existen dudas razonables sobre el momento adecuado para iniciar el tratamiento en los diagnósticos precoces que aún no han empezado con el deterioro neurológico. Se recomienda administrarlo justo en el momento en el que aparecen los síntomas neurológicos18. Esta decisión es difícil en los casos con retraso psicomotor previo como en el caso 1.
La RM cerebral en la enfermedad de NPC no suele mostrar imágenes específicas. Es frecuente ver atrofia cerebral a lo largo de la evolución y en particular atrofia del vermis cerebeloso19. También se puede observar adelgazamiento del cuerpo calloso y aumento de la señal en la sustancia blanca que puede reflejar una desmielinización secundaria15. En la enfermedad de NPC se pueden poner de manifiesto alteraciones en la espectroscopia cerebral20 (descenso de la N-acetil-aspartato/creatina (Naa/Cr) en corteza frontal y parietal, centro semioval y nucleo caudado y aumento de colina/creatina en corteza frontal y centro semioval)21. En nuestra serie, algunos casos presentaron descenso de Naa. La RM con espectroscopia puede ser útil en el seguimiento de la enfermedad. En las figuras 1 y 2 se muestran las imágenes de las RM de los pacientes.
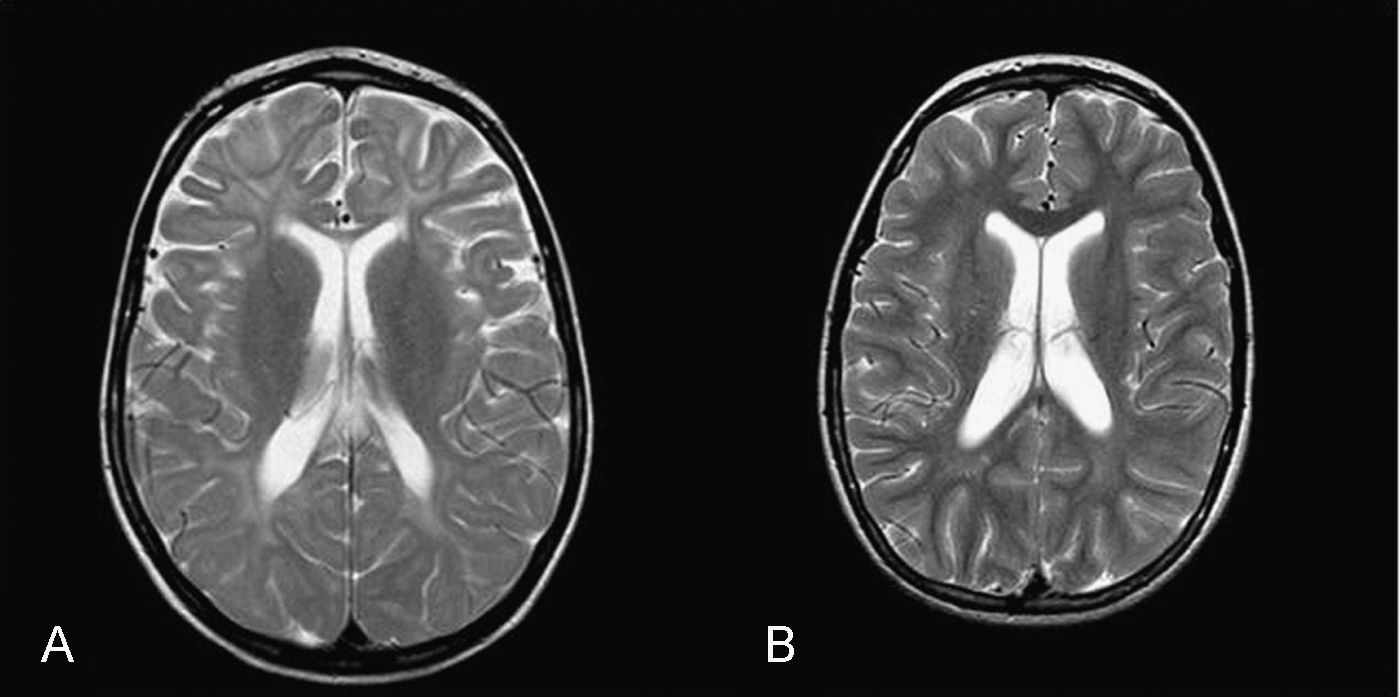
El análisis bioquímico mostró que todos los pacientes presentan el fenotipo bioquímico clásico que es el más frecuente, a excepción del paciente 4 que presentó un fenotipo bioquímico variante22. El fenotipo variante suele dar formas juveniles o adultas, sin embargo nuestro caso es una forma infantil precoz. No se han encontrado hasta el momento marcadores bioquímicos de actividad de la enfermedad18 y que además sirvan como control del tratamiento. La quitotriosidasa no ha demostrado ser un buen marcador de progresión de la enfermedad23.
El análisis molecular detectó mutaciones en el gen NPC1 en todos los casos como en la mayor parte de los casos publicados7,24. Esto permite el consejo genético a través del diagnóstico de heterocigotos y del diagnóstico prenatal. La heterogeneidad alélica de esta enfermedad es muy grande y dificulta la correlación genotipo/fenotipo clínico salvo algunas excepciones. En el caso de la mutación p.C177Y, hasta la fecha, siempre que se presentaba en homocigosis se correlacionaba con la presentación infantil tardía24. En este trabajo, presentamos el primer paciente (caso 5) con este genotipo, pero con una presentación infantil precoz (tabla 2). Por otro lado, son de destacar ciertas correlaciones entre el genotipo y el fenotipo bioquímico. Este es el caso de la mutación p.G992W, que tanto si se presenta en homo como en heterocigosis, presenta una forma bioquímica variante después de realizar la tinción citoquímica con filipina tal y como se ha descrito para todas las mutaciones que afectan al codón 99225,26.
ConclusionesLa enfermedad de NPC debe incluirse en el diagnóstico diferencial de la ascitis de origen prenatal, la colestasis neonatal y la esplenomegalia, para poder realizar un diagnóstico y un tratamiento precoces. Debemos pensar en la enfermedad de NPC en cuadros neurodegenerativos sobre todo si hay alteración de la mirada vertical. La presencia de esplenomegalia orienta el diagnóstico pero su ausencia no lo descarta. Hay que resaltar que los primeros síntomas suelen ser precoces y extraneurológicos y preceden en un tiempo variable (meses o años) al deterioro neurológico. Es imprescindible la confirmación diagnóstica mediante el estudio citoquímico utilizando filipina sobre un cultivo de fibroblastos procedentes de una biopsia de piel y/o a través del análisis molecular en los genes NPC1 y NPC2 si fuese necesario. Esto permitirá además realizar un consejo genético adecuado.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
