








Adalimumab (ADA), anticuerpo anti-TNF-α monoclonal recombinante de origen humano, generalmente se emplea como tratamiento de segunda línea en niños con enfermedad de Crohn (EC) que no han respondido o han perdido respuesta a infliximab (IFX). En las series publicadas más del 70% de los pacientes habían sido tratados inicialmente con IFX. Los datos sobre la eficacia a corto y a largo plazo de ADA en pacientes naïve a anti-TNF son muy limitados. El objetivo del presente estudio es describir nuestra experiencia con ADA como tratamiento anti-TNF de primera línea en niños con EC.
Material y métodoEstudio multicéntrico, retrospectivo que incluye pacientes con EC tratados con ADA como anti-TNF de primera línea.
ResultadosSe incluyeron 62 pacientes (34varones) con una edad media de 13,0±2,4años, un tiempo de evolución de la enfermedad de 7,3meses (RIQ 2,7-21) y un wPCDAI de 35puntos (RIQ 24,3-47,5). En el momento de comenzar ADA, 58 pacientes (93,5%) estaban recibiendo tratamiento inmunomodulador. A las 12semanas de tratamiento el 80,6% (50/62) habían alcanzado la remisión clínica, así como el 95% (57/60) a las 52 semanas. Ocho pacientes (13%) presentaron efectos adversos. Se constató un incremento significativo de los z-scores de talla, velocidad de crecimiento e índice de masa corporal (IMC) a las 52 semanas de tratamiento, en especial en aquellos con retraso de crecimiento.
ConclusionesEl tratamiento con ADA favorece una remisión clínica prolongada en pacientes naïve a anti-TNF. El tratamiento con ADA mejora la velocidad de crecimiento en niños con EC y retraso de crecimiento al inicio del tratamiento.
Adalimumab (ADA), a monoclonal humanised anti-TNF antibody, is usually prescribed as a second-line treatment in paediatric Crohn's disease (CD) patients who have become unresponsive or developed intolerance to infliximab (IFX). In the case series reported, more than 70% of patients had initially been treated with IFX. Data on short- and long-term effectiveness of ADA in anti-TNF naïve patients is limited. The aim of this study is to describe our experience with ADA as a first-line anti-TNF in paediatric CD patients.
Material and methodsThis is a multicentre retrospective study including anti-TNF naïve paediatric CD patients treated with ADA as first-line anti-TNF.
ResultsSixty-two patients (34males), with a mean age of 13.0±2.4years and a disease duration of 7.3 (IQR 2.7-21) months were included. Median wPCDAI was 35 (IQR 24.3-47.5). Fifty-eight out of 62 (93.5%) were on combo therapy at baseline. Clinical remission at week12 was achieved in 50 out of 62 (80.6%) and in 57 out of 60 (95.0%) at week52. Eight patients (13%) reported adverse events. Mean height, growth rate and BMI z-scores improved significantly between baseline and week 52, especially in patients with growth failure.
ConclusionsADA treatment leads to lasting clinical remission in anti-TNF naïve paediatric patients with CD. ADA significantly improved growth rate in children with CD who had growth delay at baseline.
La enfermedad de Crohn (EC) es una enfermedad inflamatoria intestinal crónica que puede afectar de forma segmentaria y transmural a cualquier parte del tracto digestivo. La incidencia y prevalencia de la enfermedad inflamatoria intestinal en niños se han visto incrementadas en las últimas décadas. Concretamente, la incidencia de EC en España ha pasado de 0,53 a 1,7 casos por 100.000 individuos menores de 18 años en los últimos 14 años1,2. El retraso del crecimiento y el retraso puberal son manifestaciones específicas de la edad pediátrica. El tratamiento con anti-TNF se recomienda para inducir la remisión en niños con enfermedad activa resistente a los corticoides, para inducir y mantener la remisión en niños con enfermedad luminal crónicamente activa a pesar de haber recibido tratamiento inmunomodulador optimizado (azatioprina [AZA], mercaptopurina [MP] o metotrexato [MTX]), y como tratamiento inicial de inducción y mantenimiento en niños con enfermedad fistulizante perianal en combinación con el tratamiento quirúrgico apropiado3.
Infliximab (IFX) y adalimumab (ADA) son anticuerpos monoclonales, el primero quimérico humano/murino y el segundo totalmente humano, que se unen específicamente al factor de necrosis tumoral alfa (TNFα). El IFX se ha mostrado eficaz en el tratamiento de niños con EC4, siendo utilizado, por lo general, como tratamiento anti-TNF de primera línea en niños. La indicación de ADA para el tratamiento de la EC en niños fue autorizada con posterioridad al IFX. La evidencia de la eficacia de ADA en poblaciones pediátricas viene refrendada por estudios prospectivos y retrospectivos y el ensayo clínico IMAgINE15–12. En la práctica clínica, el ADA se suele emplear en pacientes que han perdido respuesta o desarrollado intolerancia a IFX13. El ADA también ha mostrado su utilidad como tratamiento anti-TNF de primera línea en adultos. No obstante, la experiencia sobre la eficacia a largo plazo de ADA en niños con EC naive a anti-TNF es limitada14.
El objetivo del presente estudio es evaluar la efectividad y la seguridad de ADA para inducir y mantener la remisión clínica en niños con EC naive al tratamiento con fármacos anti-TNF.
Material y métodosEstudio retrospectivo multicéntrico en tres hospitales terciarios de referencia. Se revisaron los datos clínicos de todos los pacientes con EC menores de 18años que recibieron ADA como tratamiento anti-TNF de primera línea. Todos los pacientes eran naive al tratamiento anti-TNF, pero podían haber sido tratados anteriormente con nutrición enteral exclusiva, corticoides y/o inmunomoduladores (AZA, 6MP o MTX).
La EC se diagnosticó según los criterios de Oporto revisados15. Antes de iniciar el tratamiento se descartó infección por tuberculosis mediante la pruebas de Mantoux y el QuantiFERON®-TB Gold In-Tube test (Cellestis Ltd; Carnegie, VIC, Australia). El fenotipo de la EC se estableció atendiendo a la Clasificación de París16.
Previamente a la inclusión de cada caso, los autores (JMC, GPM, MNR y VMNL) lo evaluaron y debatieron la indicación del tratamiento anti-TNF: se requirió un consenso absoluto (4/4) para incluir a cada paciente en el estudio. Los criterios de exclusión fueron: presencia de infección activa; insuficiencia cardiaca, renal o hepática, y trastorno neurológico o inmunodeficiencia. El área de superficie corporal se calculó mediante la fórmula de Haycock17.
La eficacia del tratamiento se evaluó mediante el índice ponderado de actividad de la EC pediátrica (weighted Pediatric Crohn's Disease Actvity Index [wPCDAI]). La remisión se definió si el wPCDAI < 12,5 puntos, la respuesta leve como un descenso de más de 12,5 puntos con respecto al wPCDAI basal y la respuesta moderada si el descenso del wPCDAI fue superior a 37,5 puntos sobre el basal. La no respuesta primaria se definió por wPCDAI > 12,5 puntos tras el tratamiento de inducción18. Además del wPCDAI se emplearon otros criterios de remisión.
La intensificación del tratamiento se indicó en pacientes con pérdida de respuesta, definida como recurrencia clínica (wPCDAI ≥ 12,5) durante el tratamiento de mantenimiento tras haber alcanzado la remisión clínica después de la inducción19. El wPCDAI se calculó al inicio y en las semanas12 y 52. Se utilizó la calprotectina fecal (CF, Calprest®, Eurospital, Trieste, Italia) para establecer el grado de inflamación de la mucosa intestinal. Las muestras para la determinación de la CF fueron recogidas por el propio paciente en su domicilio y se entregaron refrigeradas al laboratorio para su análisis. Los niveles de CF inferiores a 50μg/g de heces se consideraron normales.
La dosis de inducción de ADA (administrada cada 2semanas) se estableció según el peso basal: 160mg y 80 mg, o 80mg y 40mg para pacientes con peso ≥40kg o <40kg, respectivamente. Las dos primeras dosis de ADA se inyectaron en el hospital bajo supervisión médica. Las dosis posteriores fueron administradas en el domicilio del paciente por los padres, o en su centro de atención primaria. La dosis de mantenimiento estándar fue de 40mg cada 2semanas. Se determinaron niveles de proteína C reactiva (PCR), velocidad de sedimentación globular (VSG), hemoglobina (Hb), hematocrito (Hct), plaquetas, leucocitos, albúmina sérica y CF durante el seguimiento. Los niveles de PCR también se expresaron como el cociente del valor obtenido y el límite superior de la normalidad (LSN). El peso y la talla se midieron con el paciente descalzo y en ropa interior. Los z-score de peso, talla, índice de masa corporal (IMC) y velocidad de crecimiento se calcularon empleando como referencia las curvas de crecimiento en la población española20. El retraso de crecimiento grave se definió como un z-score de velocidad de crecimiento por debajo de −2,5, y el retraso leve-moderado como un z-score de velocidad de crecimiento entre −1,0 y −2,53. En los casos en los que existía retraso puberal la velocidad de crecimiento se calculó en base al estadio de Tanner y la edad ósea. Se definió como efecto adverso grave cualquier efecto o reacción adversa potencialmente letal, que ocasionara la muerte o condicionara hospitalización o prolongación de la hospitalización en curso, o que desembocase en discapacidad o invalidez persistente o significativa.
En un primer análisis se estudió la remisión libre de corticoides en las semanas12 y 52. En segundo lugar se analizó la necesidad de intensificación y el efecto de la terapia anti-TNF con ADA sobre la velocidad de crecimiento.
Análisis estadísticoLas variables que seguían una distribución normal se expresaron como media ± desviación estándar (DE) y las variables con distribuciones no normales como mediana y rango intercuartílico (RIQ). La normalidad de la distribución de los datos se comprobó mediante la prueba de Kolmogorov-Smirnov. Se utilizaron la prueba de la t de Student y la prueba de los rangos con signo de Wilcoxon para muestras pareadas, y la prueba de chi-cuadrado para comparar proporciones. Se empleó la regresión logística para determinar los predictores de respuesta e intensificación. El nivel de significación estadística se estableció en p<0,05.
Consideraciones éticasSe obtuvo consentimiento informado por parte de los pacientes o sus padres. El estudio fue aprobado por los comités éticos de todos los centros participantes.
ResultadosCaracterísticas demográficasSe incluyeron 62 niños (34 varones) con EC y naive al tratamiento anti-TNF. Las características demográficas y clínicas basales de los pacientes se muestran en la tabla 1. En cuanto a la localización de la enfermedad, 31 (50%) presentaban afectación ileocólica (L3) y 18 (29%), extensa (L3L4a/b). La presentación fenotípica más frecuente fue el patrón no estenosante-no penetrante (B1, 77,4%). Se observó retraso de crecimiento (G1) en 24 pacientes (38,7%). Casi todos los pacientes recibieron NEE (52 [84%]) y tiopurinas (56 [90%]) antes de iniciar tratamiento con ADA. Diez pacientes (16%) habían recibido tratamiento previo con corticoides. El ADA se indicó tras el fracaso del tratamiento de inducción con NEE (n=4 [6,5%]), el fracaso de la terapia de mantenimiento con AZA tras el éxito de la NEE (n=41 [66%]), debido a la presencia de manifestaciones extraintestinales graves (n=7 [11,3%]), afección perianal grave (n=4 [6,5%]), como estrategia tipo top-down para el tratamiento de enfermedad grave (n=3 [4,8%]) o por intolerancia al tratamiento previo (AZA o MTX, n=6 [9,6%]).
Características basales de los pacientes tratados con adalimumab
| Varón | 34/62 (55%) | |
| Edad al diagnóstico (años) | 11,6±3,1 | |
| Tiempo transcurrido hasta diagnóstico (meses) | 5,2 (RIQ 2,8-12,6) | |
| Edad inicio de adalimumab (años) | 13,0±2,4 | |
| Duración de la enfermedad (meses) | 7,3 (RIQ 2,7-21) | |
| Dosis de inducción con adalimumab | ||
| 160/80 | 41 (66%) | |
| 80/40 | 21 (34%) | |
| Localización (París) | ||
| L1 | 6 (9,7%) | |
| L2 | 1 (1,6%) | |
| L3 | 31 (50%) | |
| L4 | 1 (1,6%) | |
| L1+L4 | 3 (4,8%) | |
| L2+L4 | 2 (3,2%) | |
| L3+L4 (extensa) | 18 (29%) | |
| B1 | 48 (77,4%) | |
| B2 | 10 (16,1%) | |
| B3 | 4 (6,5%) | |
| Afección perianal (p) | 13 (21%) | |
| Retraso de crecimiento (G1) | 20 (32,2%) | |
| Enfermedad extraintestinal | 5 (8%) | |
| wPCDAI basal | 35 (RIQ 24,3-47,5)a | |
| Tratamiento previo | ||
| Nutrición enteral exclusiva | 52 (84%) | |
| Corticoides | 10 (16%) | |
| Aminosalicilatos | 8 (13%) | |
| Azatioprina | 53 (85,5%) | |
| Mercaptopurina | 3 (4,8%) | |
| Budesonida | 6 (9,6%) | |
| Metotrexato | 2 (3,2%) | |
| Tratamiento al inicio de adalimumab | ||
| Nutrición enteral exclusiva | 5 (8%) | |
| Corticoides | 3 (4,8%) | |
| Azatioprina/Mercaptopurina | 57 (92%) | |
| Metotrexato | 1 (1,6%) | |
| Dosis medias de adalimumab durante la inducción y el mantenimiento | mg/kg | mg/m2 |
| 1.a dosis | 3,3±0,9 | 101±25,8 |
| 2.a dosis | 1,6±0,5 | 50,6±12,9 |
| Dosis acumulada hasta semana 12 | 9,2±2,5 | 278,2±49,0 |
| Dosis mantenimiento a las 12 semanas | 0,98±0,33 | 30,4±6,5 |
| Dosis mantenimiento a las 52 semanas | 0,83±0,25 | 27,5±5,3 |
| Cambio de dosis mantenimiento entre semanas 12 y 52 | 0,11 (RIQ 0,04-0,2) | 2,3 (RIQ 1,0-3,9) |
| Parámetros analíticos basales | ||
| Calprotectina fecal (μg/g) | 749 (RIQ 428-1077) | |
| PCR (mg/L) | 27 (RIQ 9,8-59) | |
| PCR/LSN (mg/L) | 2,9 (RIQ 1,0-5,4) | |
| PCR normal | 16/62 (26%) | |
| VSG (mm/h) | 21 (RIQ 14-35,5) | |
| Albúmina (g/dl) | 3,9 (RIQ 3,6-4,2) | |
| Hb (g/dl) | 12,1 (RIQ 11,0-13,0) | |
| Hct (%) | 37 (RIQ 35,3-39,1) | |
| Leucocitos (×109/L) | 7,57 (RIQ 5,92-10,65) | |
| Plaquetas (×109/L) | 392 (RIQ 331-477) | |
Hb: hemoglobina; Hct: hematocrito; LSN: límite superior de la normalidad; PCR: proteína C reactiva; RIQ: rango intercuartílico; VSG: velocidad de sedimentación globular; wPCDAI: weighted paediatric Crohn's disease activity index.
En el curso del período de estudio (2008-2015) 89 pacientes con EC fueron tratados con anti-TNF en los tres centros, 62 recibieron ADA (69%) y 27 IFX (31%). Cincuenta y seis de los 62 pacientes o sus padres eligieron ADA como terapia anti-TNF en comparación con 5 de los 27 tratados con IFX (90,3% vs. 18,5%; p<0,0001). En 16 casos tratados con IFX y 6 tratados con ADA, estos biológicos se seleccionaron por diferentes motivos (criterios médicos, problemas sociales, adherencia al tratamiento, etc.) sin dar opción al paciente.
Pautas de inducción y mantenimiento con adalimumabLas dosis utilizadas para la inducción y el mantenimiento de la remisión se muestran en la tabla 1. Las dosis de mantenimiento en la semana52 fueron menores que las de la semana12 (p<0,0001) debido exclusivamente al incremento del peso de los pacientes.
Remisión y respuesta clínicaTranscurridas 12 semanas de tratamiento, 50 de los 62 pacientes habían alcanzado la remisión clínica (80,6%), mientras que 3 pacientes mostraron una respuesta leve (tabla 2). Ni los valores de wPCDAI, PCR, CF y VSG, ni la dosis de inducción por kilogramo o por metro cuadrado de superficie corporal en las primeras 12semanas de tratamiento con ADA o la dosis total de ADA hasta la semana12 por kilogramo o por metro cuadrado de superficie corporal sirvieron como predictores de respuesta a las 12semanas. No se solicitaron niveles del fármaco ni de anticuerpos.
Tasas de remisión a las 12 y 52 semanas
| Criterios de remisión | 12 semanas | Necesidad de intensificacióna | 52 semanas | |
|---|---|---|---|---|
| % Remisiónb | n (%) | % Remisión | ||
| PPc | ITT | |||
| wPCDAI<12,5 | 50/62 (80,6%) | 4/49 (8,1%) | 57/60 (95,0%) | 57/62 (91,9%) |
| wPCDAI<12,5 y VSG<20mm/h | 43/62 (69,8%) | 2/43 (4,6%) | 49/60 (81,6%) | 49/62 (79,0%) |
| wPCDAI <12,5 y VSG<20mm/h y PCR<5mg/L | 27/51 (52,9%) | 1/27 (3,7%) | 40/55 (72,7%) | 40/62 (64,5%)d |
| wPCDAI <12,5 y VSG <20mm/h y PCR/LSN<1mg/L | 33/51 (82,3%) | 1/33 (3,0%) | 46/55 (83,6%) | 46/62 (74,2%)d |
| wPCDAI<12,5 y PCR<5mg/L | 33/56 (58,9%) | 3/33 (9,1%) | 44/55 (80,0%) | 44/62 (70,9%)d |
| wPCDAI<12,5 y PCR/LSN<1mg/L | 40/56 (71,4%) | 3/40 (7,5%) | 50/55 (90,9%) | 50/62 (80,6%)d |
| wPCDAI<12,5 y CF<250μg/g | 24/39 (61,5%) | 2/24 (8,3%) | 16/21 (76,2%) | 16/62 (25,8%)d |
| wPCDAI<12,5, PCR <5mg/L y CF<250μg/g | 12/34 (35,3%) | 1/12 (8,3%) | 15/24 (44,8%) | 15/62 (24,2%)d |
| wPCDAI<12,5, PCR/LSN≤1mg/L y CF<250μg/g | 12/34 (35,3%) | 1/12 (8,3%) | 11/31 (35,5%) | 11/62 (17,4%)d |
| wPCDAI=0, PCR<5mg/L y CF<250μg/g | 11/37 (29,7%) | 1/11 (9,1%) | 11/31 (35,5%) | 11/62 (17,4%)d |
| wPCDAI=0, PCR/LSN≤1mg/L y CF<250μg/g | 11/37 (29,7%) | 1/11 (9,1%) | 13/23 (56,5%) | 13/62 (20,9%)d |
CF: calprotectina fecal; LSN: límite superior de la normalidad; PCR: proteína C reactiva; VSG: velocidad de sedimentación globular; wPCDAI: weighted paediatric Crohn's disease activity index.
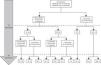
A las 52 semanas, 57 de los 60 pacientes (95%, análisis PP) habían alcanzado la remisión clínica (tabla 2) y 2 habían interrumpido el tratamiento. De estos 57 pacientes, 10 (17,5%) precisaron intensificación (40 mg/semanales) durante el primer año de tratamiento, con una duración mediana de 6meses (RIQ 2,2-10,3). De todos los pacientes que requirieron intensificación durante el primer año de tratamiento, el 66,7% habían alcanzado la remisión clínica a las 52semanas (fig. 1).
Terapia combinada en comparación con monoterapia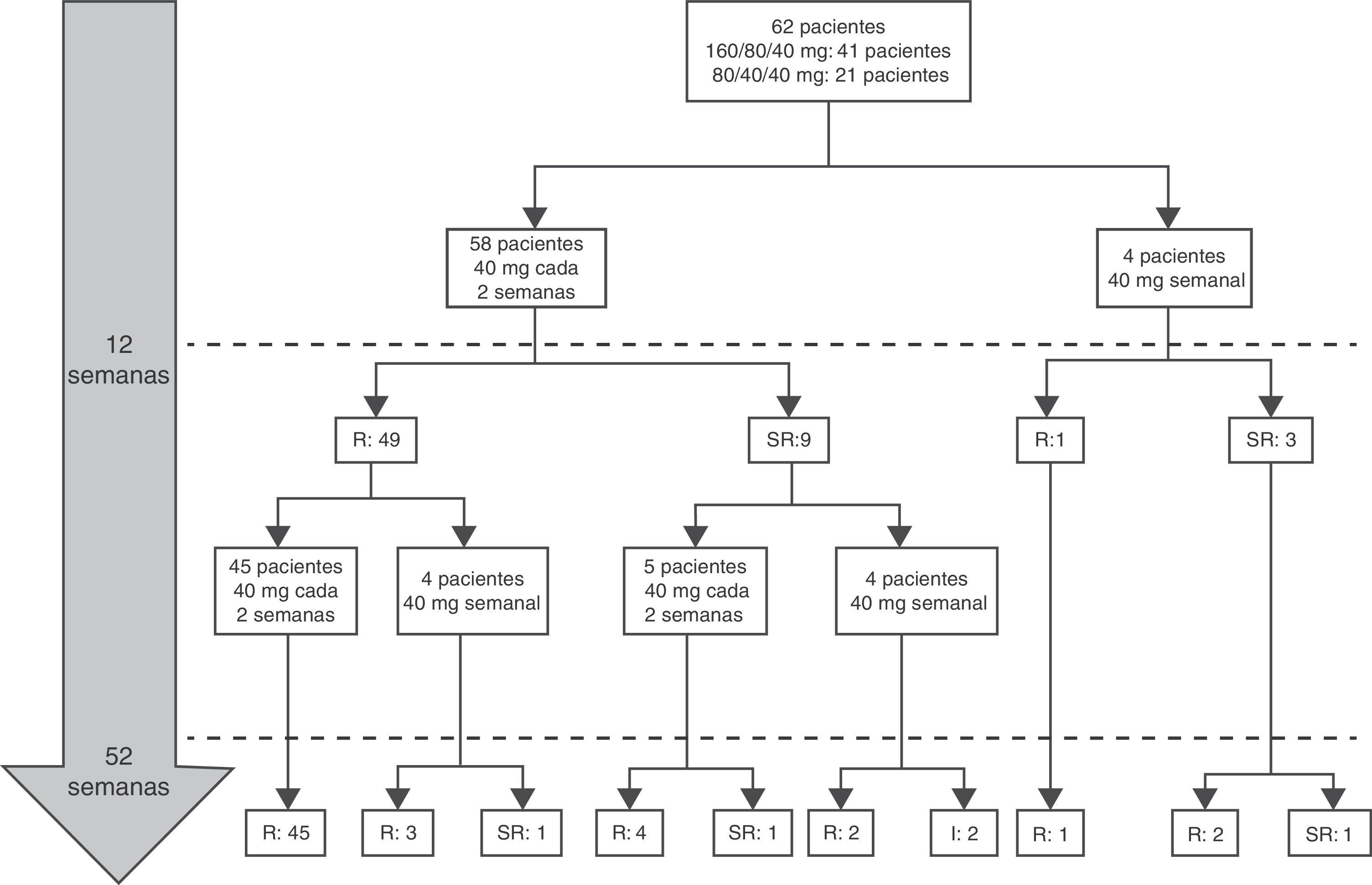
Al inicio, 58 de los 62 pacientes (93,5%) recibían terapia combinada (tabla 1). Sin embargo, a los 12meses de seguimiento solo 23pacientes (14niñas) recibían terapia combinada, mientras que 39 (63%) no la recibían a los 7meses (RIQ 4-11). La duración mediana de la terapia combinada fue de 12meses (RIQ 6-14). En 10pacientes se retiró el ADA a los 8,1meses (RIQ 6,5-20,2). En 2pacientes el tratamiento con ADA se interrumpió a partir de la semana12, en un caso por fallo primario (niveles valle de ADA>12μg/ml y anticuerpos negativos) y en el otro por desarrollo de anticuerpos anti-ADA, requiriendo cambio a IFX, con buena respuesta. Los motivos para la retirada de ADA fueron remisión clínica durante mantenimiento con monoterapia (7 casos); efectos adversos (1 caso), y fallo terapéutico (2 casos).
IntensificaciónLa intensificación fue necesaria en 16 de los 62casos (25,8%), en 12 (75%) durante el primer año de tratamiento a una mediana de 12,9 semanas (RIQ 8,4-17,7) tras el inicio de ADA. Doce de 16pacientes (75%) recibían terapia combinada en el momento de la escalada. De ellos, 11 (68,7%) habían regresado a la pauta estándar al final del período de seguimiento. La mediana del tiempo durante el cual estos pacientes requirieron tratamiento semanal con ADA fue de 5,9meses (RIQ 2,1-10,5). Ninguna de las características basales de los pacientes fue predictiva de la necesidad de intensificación en las primeras 12semanas de tratamiento. Sin embargo, los criterios de remisión a las 12 semanas sí anticipaban la intensificación del tratamiento en las 40 semanas siguientes. El criterio de remisión que mejor predijo la no necesidad de intensificación en el primer año de tratamiento fue un wPCDAI<12,5, una VSG<20mm/h y un cociente PCR/LSN<1 (OR 0,048 [0,005-0,4550], p=0,008). El que tuvo peor capacidad predictiva fue el wPCDAI de forma aislada (fig. 2).
Efectos adversos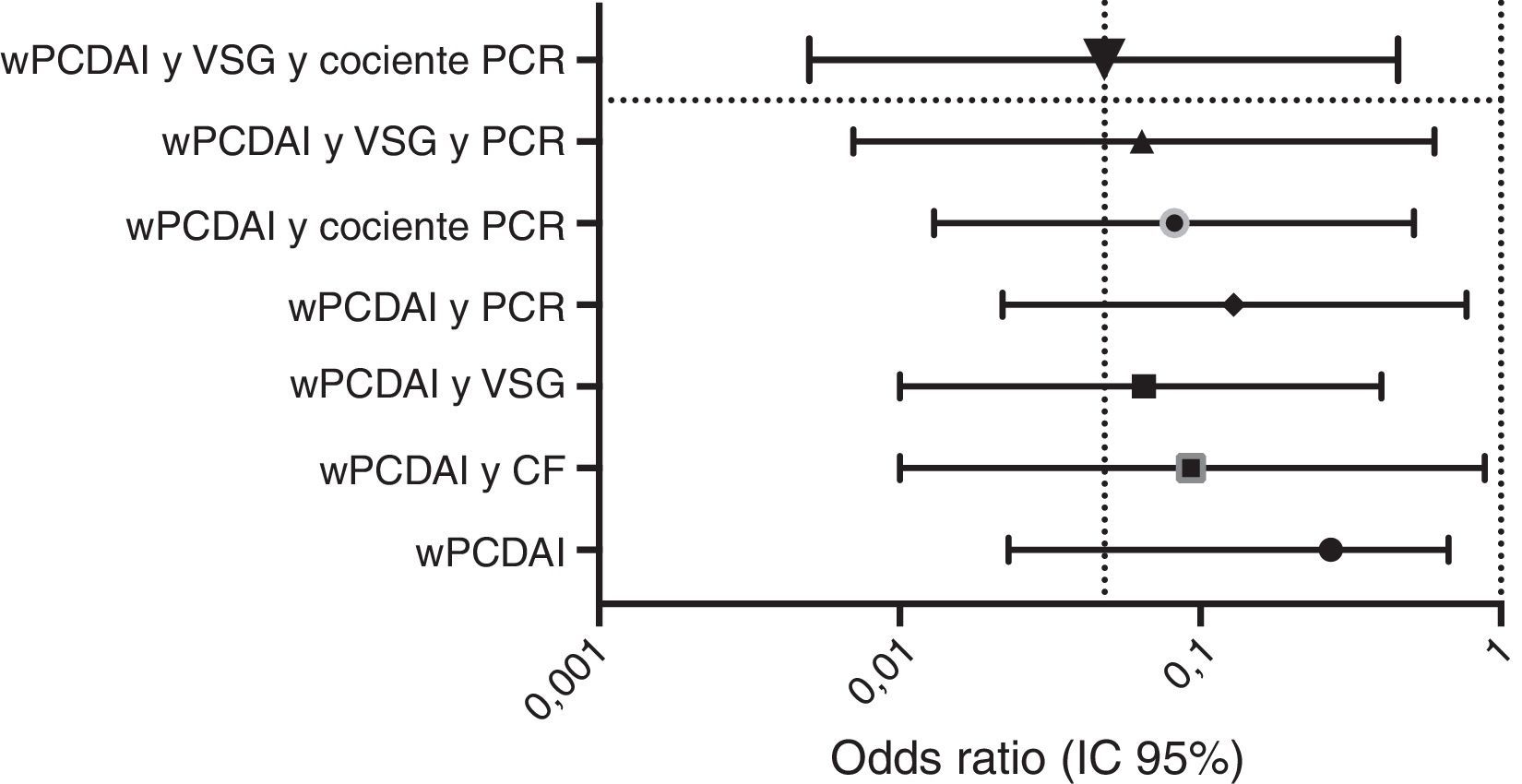
Se inyectaron un total de 3.103 dosis, con una duración mediana de tratamiento de 15,7meses (RIQ 8,1-33), una mediana de 42dosis por paciente (RIQ 30-61) y una duración mediana de seguimiento de 20,7meses (RIQ 14,2-33,5). Ocho pacientes (13%) reportaron efectos adversos: episodios recurrentes de amaurosis fugax (1), psoriasis (1), temblores (1), síndrome depresivo (1) y reacciones leves en el lugar de la inyección. No se consignaron reacciones alérgicas, infecciones graves tales como tuberculosis u otras infecciones oportunistas, o malignidad.
Cambios en marcadores analíticos durante el tratamientoLos niveles y el cociente de PCR, la VSG, el recuento leucocitario, el recuento de plaquetas y los niveles de CF habían descendido de manera significativa a las 12 y 52semanas, constatándose por otro lado aumentos significativos en las cifras de hemoglobina, el hematocrito y la albúmina sérica (tabla 3). Se recogieron valores de CF en 23 pacientes a las 52 semanas; los niveles de CF eran inferiores a 250μg/g en 12 (52%) e inferiores a 50μg/g en 8 (35%).
Evolución del wPCDAI y parámetros analíticos tras el tratamiento con adalimumab
| Inicio | 12 semanas | 52 semanas | p | |||
|---|---|---|---|---|---|---|
| Semanas 0-12a | Semanas 12-52a | Semanas 0-52a | ||||
| wPCDAI | 35 (RIQ 24,3-47,5) | 0 (RIQ 0-7,5) | 0 (RIQ 0-0) | 0,0001 | 0,026 | 0,0001 |
| CF (μg/g) | 749 (RIQ 428-1.077) | 140 (RIQ 49,7-281) | 92 (RIQ 25-366) | 0,0001 | 0,681 | 0,0001 |
| PCR (mg/L) | 27 (RIQ 9,8-59,7) | 2 (RIQ 0,5-5) | 1,0 (RIQ 0,5-3,2) | 0,0001 | 0,108 | 0,0001 |
| PCR/LSN | 2,8 (RIQ 1,0-5,4) | 0,16 (RIQ 0,06-0,51) | 0,09 (RIQ 0,06-0,33) | 0,0001 | 0,075 | 0,0001 |
| VSG (mm/h) | 21 (RIQ 14-35) | 7 (RIQ 3-15) | 6 (RIQ 3-11) | 0,0001 | 0,142 | 0,0001 |
| Albúmina (g/dl) | 3,9 (RIQ 3,6-4,2) | 4,2 (RIQ 3,9-4,5) | 4,4 (RIQ 4,2-4,5) | 0,0001 | 0,024 | 0,0001 |
| Hb (g/dl) | 12,0 (RIQ 10,9-13,0) | 12,6 (RIQ 11,8-13,6) | 13,3 (RIQ 12,5-14,1) | 0,0001 | 0,0001 | 0,0001 |
| Hct (%) | 37 (RIQ 35,3-39,1) | 38,4 (RIQ 36,6-41,2) | 40,4 (RIQ 38,1-42,1) | 0,001 | 0,0001 | 0,0001 |
| Plaquetas (×103/μl) | 392 (RIQ 331-477) | 296 (RIQ 262-360) | 286 (RIQ 230-339) | 0,0001 | 0,004 | 0,0001 |
| Leucocitos (×103/μl) | 7.575 (RIQ 5.925-10.665) | 6.400 (RIQ 4.800-7.545) | 6.175 (RIQ 5.057-7.865) | 0,0001 | 0,0001 | 0,907 |
La velocidad de crecimiento y el IMC se midieron al inicio de tratamiento con ADA y a las 52semanas de seguimiento. Las medias de los z-scores de talla, velocidad de crecimiento e IMC mejoraron significativamente entre el inicio y la semana52 (fig. 3). Como se observa en la figura 4, el tratamiento con ADA mejoró significativamente el z-score de la velocidad de crecimiento en niños con EC, especialmente en aquellos con fallo de crecimiento grave al inicio.
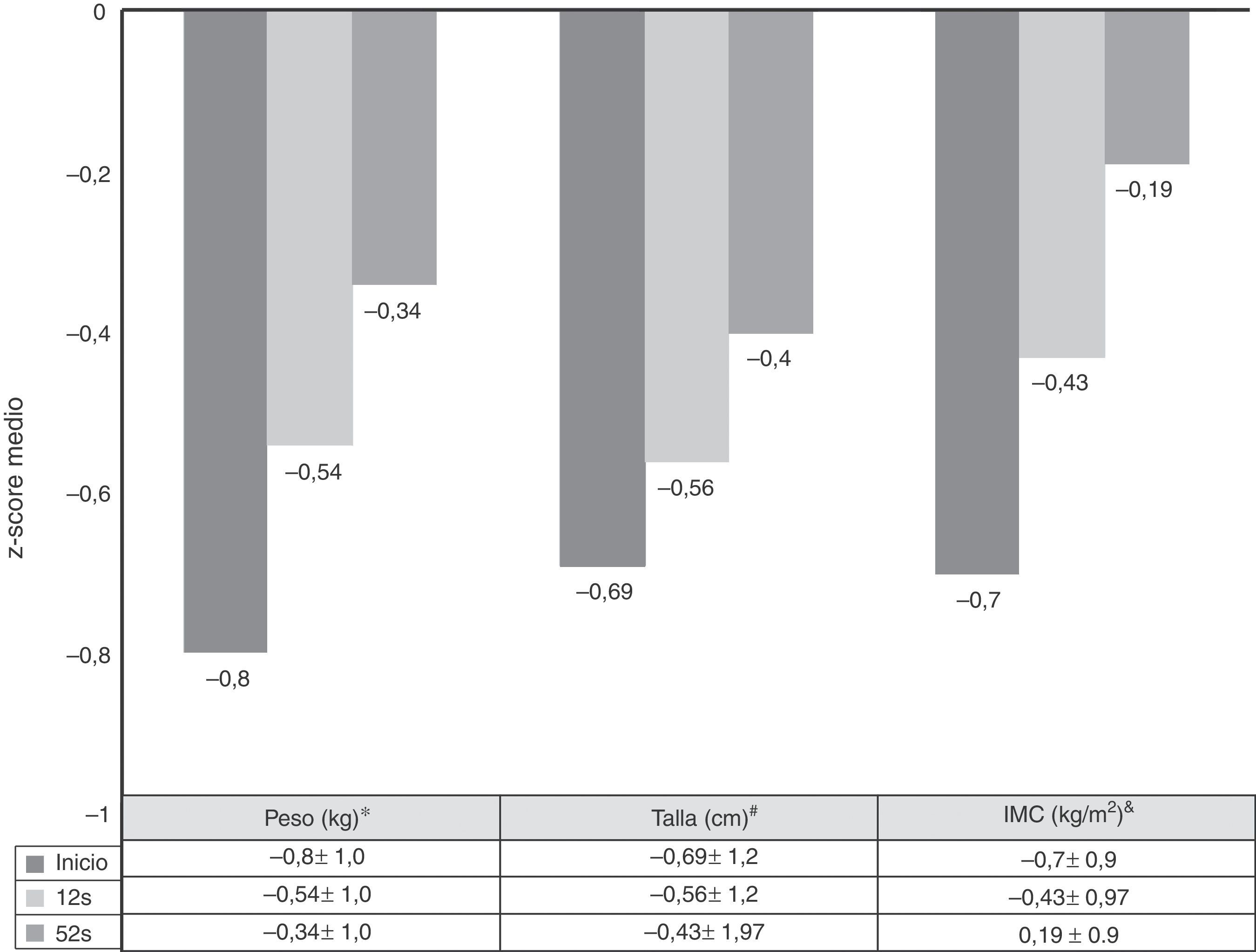
Evolución de los z-scores de peso, talla e IMC desde el inicio hasta las 52 semanas.
* Inicio vs 12 semanas: p=0,0001; 12 vs 52 semanas: p=0,01; inicio vs 52 semanas: p=0,0001.
# Inicio vs 12 semanas: p=0,005; 12 vs 52 semanas: p=0,0001; inicio vs 52 semanas: p=0.0001.
& Inicio vs 12 semanas: p=0,0001; 12 vs 52 semanas: p=0,804; inicio vs 52 semanas: p=0,039.
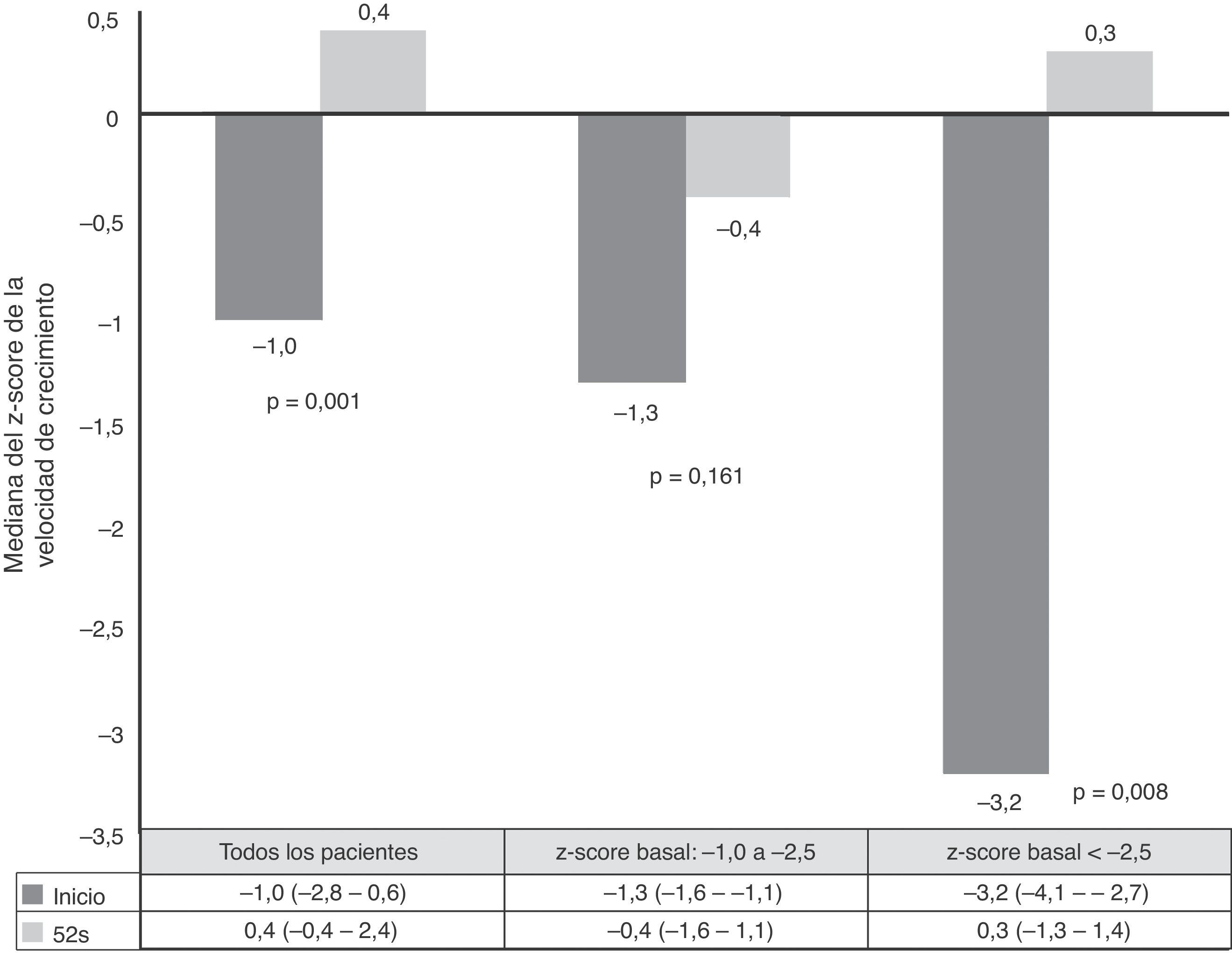
La literatura publicada en torno al tratamiento con ADA en pacientes pediátricos con EC naïve a anti-TNF es muy escasa21. Nuestros datos muestran resultados prometedores en efectividad, seguridad y crecimiento en base a las tasas de remisión clínica a las 12 y 52semanas de iniciar el tratamiento. Un estudio realizado por Hyams et al.10, que incluyó a 105pacientes naive a IFX, mostró una tasa de remisión del 56,9% a las 26semanas y del 45,1% a las 52semanas en el grupo con dosis alta (40mg cada 2semanas) en comparación con el 35,2% a las 26semanas y el 27,8% a las 52semanas en el grupo con dosis baja (20mg cada 2semanas), lo que constituyó el principal motivo de que no se empleasen dosis bajas de ADA en nuestro estudio. Otra posible explicación de nuestros mejores resultados sería el menor tiempo de evolución antes de la dosis inicial de ADA (mediana de 7,3meses en nuestro estudio versus 2,9años en el estudio de Hyams). En un estudio prospectivo reciente que incluía a 12 pacientes naïve a anti-TNF tratados con ADA con un tiempo medio de evolución de enfermedad de 9,4±2,7meses (rango 8,6-15,7), la tasa de remisión a los 9-12meses de tratamiento con ADA fue del 67,5%, con curación mucosa completa en el 41,7% de los pacientes y curación mucosa parcial en el 25%12. Varios estudios han constatado una relación inversa entre el tiempo de evolución de la enfermedad y los resultados del tratamiento21,22.
Los resultados de nuestro estudio aportan evidencia adicional en apoyo de la hipótesis de que ADA puede dar mejores resultados en pacientes naïve a anti-TNF que en pacientes previamente tratados con IFX. Hasta ahora solo ha habido un estudio en la población pediátrica, el IMAgINE1 (que incluye el subgrupo de pacientes naïve a anti-TNF de mayor tamaño hasta la fecha) y dos ensayos en pacientes adultos, CHARM23 y CLASSICI24, que mostraron resultados positivos. Dos estudios prospectivos y varios retrospectivos centrados en la eficacia y efectividad de ADA en poblaciones pediátricas incluían una mayoría de pacientes con fallo previo de terapia anti-TNF5–7,9,21,22. La pauta de inducción de ADA en nuestra serie fue de 160/80mg o de 80/40mg en pacientes con peso ≥40kg o<40kg, respectivamente. La pauta de mantenimiento fue de 40mg cada 2semanas en todos los pacientes. La pauta empleada fue similar a la empleada en el grupo de dosis alta de ADA en el estudio de Hyams, que mostró mayor eficacia en comparación con el grupo con dosis baja10.
La duración mediana de la terapia combinada en nuestra serie fue de 12meses (RIQ 6-14). Aunque los datos sobre este aspecto en la población pediátrica son escasos, es posible que la terapia combinada durante los primeros meses de tratamiento anti-TNF se asocie a una menor tasa de desarrollo de anticuerpos y de pérdida de respuesta. No obstante, esta ventaja ha de sopesarse con el potencial aumento del riesgo de linfoma asociado a las tiopurinas25.
La malnutrición y el fallo de crecimiento son consecuencias negativas en niños con EC. Varios estudios han reportado un efecto favorable de la terapia anti-TNF sobre este aspecto26–37. No obstante, otros estudios no encontraron cambios en el crecimiento lineal26,30,31. Uno de ellos describió efectos nutricionales y sobre el desarrollo del ADA en pacientes pediátricos con EC, pero en este caso todos los pacientes habían sido tratados previamente con IFX33. Nuestro estudio es el primero en presentar datos sobre los efectos de ADA en parámetros de crecimiento y nutricionales en pacientes naïve a anti-TNF. Además, confirma su efecto beneficioso en la mejora del peso, talla, IMC y velocidad de crecimiento en un período de un año. En nuestra serie observamos mejoras en los z-scores de velocidad de crecimiento en pacientes con fallo de crecimiento al inicio. Estos resultados son similares a los publicados previamente10,26. Una posible explicación de estos resultados es el protocolo con ahorro de corticoides seguido en nuestro estudio; solo 2pacientes recibieron corticoides concomitantes con ADA por manifestaciones extraintestinales graves.
Los biomarcadores en sangre juegan un papel importante en el seguimiento de pacientes pediátricos con EC. Aunque no son tan sensibles o específicos como los biomarcadores fecales, son de gran utilidad, en combinación con las manifestaciones clínicas, para predecir la inflamación de la mucosa35. También se han descrito efectos de ADA sobre la hemoglobina, el hematocrito, el recuento leucocitario, las plaquetas y los niveles de albúmina. Todos los parámetros mostraron una normalización lenta pero progresiva, con mantenimiento de niveles normales hasta el final del seguimiento. En cuanto a la seguridad, nuestra experiencia mostró una tasa reducida de efectos adversos graves, sin casos de malignidad, infecciones oportunistas o necesidad de intervención quirúrgica. Estos hallazgos concuerdan con los de otros estudios5–7,9,26. En cambio, dos estudios previos encontraron una incidencia de reacciones en el lugar de inyección del 10-43% y una incidencia de efectos adversos del 20%10,12.
En nuestra serie, 12 de los 62 pacientes (19,3%) precisaron intensificación a 40mg de ADA a la semana durante el primer año de tratamiento, una proporción muy inferior a la publicada recientemente por Dubinsky et al.38. En dicha serie (dentro del estudio IMAgINE1), el 44% requirieron intensificación a las 12semanas de tratamiento, correspondiendo al 50,5% del grupo con dosis baja de ADA y el 37,6% del grupo con dosis alta. A las 52semanas de seguimiento el 18,8% del grupo con dosis baja y el 31,4% del grupo con dosis alta habían alcanzado la remisión. Las proporciones fueron mayores en pacientes naïve a anti-TNF tratados con dosis altas (15,4 y 44,4%), que aun así fueron inferiores a las alcanzadas en nuestra serie.
Una contribución no descrita previamente en series pediátricas fue la preferencia de los niños y sus familias de recibir inyecciones subcutáneas de ADA en el domicilio en lugar de tratamiento con IFX en el hospital. Tal y como especificaban las guías de consenso de la European Crohn's and Colitis Organisation y la European Society for Pediatric Gastroenterology and Nutrition (ECCO-ESPGHAN) sobre el tratamiento de la EC pediátrica, dado que IFX y ADA han mostrado un perfil de seguridad y eficacia similares en el tratamiento de pacientes naive a anti-TNF, ambos se deberían ofrecer como opciones a dichos pacientes en base a su disponibilidad, vía de administración, preferencia del paciente, coste y normativa local. Varias series de pacientes adultos con enfermedad subsidiaria de tratamiento anti-TNF demuestran que la facilidad de uso y el tiempo requerido para la administración son los dos factores que más influyen en la elección de tratamiento39,40. Por añadidura, la implicación del paciente y sus cuidadores en la selección del tratamiento podría reforzar no solo la relación médico-paciente, sino también la responsabilidad del paciente en su tratamiento y autocuidado. La puesta en práctica de esta estrategia en nuestros centros ha mostrado que el 90% de nuestros pacientes a los que se les dio la opción prefirieron ADA.
Este estudio tiene algunas limitaciones, incluyendo un tamaño muestral reducido, su diseño retrospectivo, la falta de un grupo de control, la corta duración del seguimiento y el no haberse realizado mediciones de niveles del fármaco en sangre o monitorización de la curación mucosa mediante colonoscopia. Aun así, esta serie aporta datos importantes: la efectividad de ADA en la normalización de la velocidad de crecimiento en pacientes pediátricos con EC y los factores predictores de intensificación de tratamiento.
En conclusión, el uso de ADA como agente anti-TNF de primera línea induce y mantiene la remisión clínica en pacientes pediátricos con EC. Además, ADA tiene un efecto beneficioso en parámetros nutricionales y de crecimiento, y es bien tolerado. Algunos pacientes en tratamiento con monoterapia mantienen remisión de forma prolongada; no obstante, algunos de ellos requieren intensificación durante el seguimiento.
Aprobación éticaTodos los procedimientos realizados en estudios con participantes humanos cumplieron las normas éticas de la institución o comité nacional de investigación y la declaración de Helsinki y sus enmiendas, o principios éticos comparables.
Consentimiento informadoSe obtuvo el consentimiento informado de cada uno de los participantes en el estudio.
AutoríaLos autores contribuyeron significativamente en:
- •
La concepción y el diseño del estudio o la adquisición o análisis e interpretación de datos: Víctor Manuel Navas-López, Gemma Pujol Muncunill, Enrique Llerena, María Navalón Rubio y Javier Martín de Carpi.
- •
La redacción del borrador del manuscrito o revisión crítica de su contenido intelectual: Víctor Manuel Navas-López, Gemma Pujol Muncunill, Enrique Llerena, María Navalón Rubio, David Gil-Ortega, Vicente Varea-Calderón, Carlos Sierra Salinas y Javier Martín de Carpi.
- •
La aprobación de la versión final a remitir: Víctor Manuel Navas-López, Gemma Pujol Muncunill, Enrique Llerena, María Navalón Rubio, David Gil-Ortega, Vicente Varea-Calderón, Carlos Sierra Salinas y Javier Martín de Carpi.
El Dr. Víctor Manuel Navas-López ha recibido honorarios por actuar como asesor y como ponente para Abbvie.
La Dra. María Navalón Rubio ha recibido honorarios por actuar como asesor y como ponente para Abbvie.
El Dr. Enrique Llerena declara no tener conflicto de intereses.
La Dra. Gemma Pujol declara no tener conflicto de intereses.
El Dr. David Gil-Ortega declara no tener conflicto de intereses.
El Dr. Vicente Varea-Calderón declara no tener conflicto de intereses.
El Dr. Carlos Sierra declara no tener conflicto de intereses.
El Dr. Javier Martín de Carpi ha recibido honorarios por actuar como asesor y como ponente para Abbvie.
Los autores agradecen a Pablo Vivanco Jódar (PhD, Meisys) por su ayuda en la redacción del manuscrito.
