



Mujer que nace tras una gestación de 37 semanas sin incidencias. Se practica cesárea por desproporción pelvicocefálica. Presenta ausencia de clavículas, dehiscencia de la sutura sagital, fontanela anterior grande y prominencia occipital. Se solicita radiografía de tórax, ecografía cerebral y abdominal y cariotipo; se halla una alteración en la primera, en la que se observa agenesia clavicular y tórax acampanado. Se diagnostica de disostosis cleidocraneal y se inicia el estudio de la familia. Hay 10 miembros afectados pertenecientes a cinco generaciones. Es constante la ausencia parcial o total de clavículas, el cierre tardío de suturas craneales y fontanelas amplias, la diástasis de sínfisis pubiana y el tórax acampanado durante los primeros días de vida. Cuatro presentan facies característica y tres, retraso en la aparición de la primera dentición, precisando exodoncia. Existe hipoacusia en tres parientes. Los tres partos documentados fueron cesárea por desproporción pelvicocefálica. Otras alteraciones son retraso pondoestatural, coxa vara y artrosis precoz.
A female who was born after a gestation of 37 weeks without incident. A caesarean was performed due to cephalopelvic disproportion. She had no clavicles, dehiscence of the sagittal suture, large anterior fontanelle and occipital prominence. A chest x-ray, abdominal and brain ultrasound and karyotype, was requested, with changes only seen in the x-ray,, which showe clavicular agenesis and flared chest. Cleidocranial dysostosis was diagnosed a family study was started. There are ten affected members in five generations. The partial or total absence of clavicles, the late closure of cranial sutures and fontanelles, the diastasis of the pubic symphysis and the flared chest during the first days of life, are constant features. Four presented characteristic facies and three delayed onset of first teeth which required extractions. There is hearing loss in three of them. The three documented childbirths were caesarean due to cephalopelvic disproportion. Other alterations are failure to thrive, coxa vara and early osteoarthritis.
La disostosis cleidocraneal es una entidad poco frecuente, que sigue un patrón de herencia autosómico dominante con expresividad variable1 y en la que es común la afectación de varios miembros de una misma familia. Se define por una serie de alteraciones típicas1–4, entre las que se encuentran la ausencia total o parcial de clavículas, alteraciones óseas craneales, facies característica (hipertelorismo, seudoprognatismo), malformaciones dentales, hipoacusia de transmisión, etc. Presentamos una familia con 11 miembros afectados de disostosis cleidocraneal, abarcando cinco generaciones.
OBSERVACIÓN CLÍNICAMujer que nace tras una gestación de 37 semanas que transcurrió sin incidencias. Se practicó una cesárea por desproporción pelvicocefálica. El peso al nacer fue de 3.070g (percentil 25); la talla, 49cm (percentil 50), y el perímetro cefálico, 33,5cm (percentil 25–50).
En una primera exploración física se observa ausencia de clavículas, dehiscencia importante de la sutura sagital, fontanela anterior de gran tamaño (3 × 3cm) y prominencia occipital. El resto de la exploración fue normal, incluyendo los reflejos arcaicos.
Presentado en el XIX Memorial Guillermo Arce y Ernesto Sánchez Villares. Salamanca, 20 y 21 de octubre de 2006.
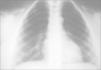
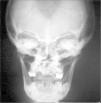
Los exámenes complementarios realizados demostraron ecografía cerebral y abdominal y cariotipo en sangre normales; radiografías craneales, en las que se aprecia la prominencia occipital, y la radiografía de tórax, en la que se constata la agenesia clavicular y se observa la imagen de tórax acampanado.
Según los hallazgos clínicos y radiológicos, se realiza el diagnóstico de disostosis cleidocraneal.
Tras el diagnóstico se inicia el estudio de la familia para comprobar si existen otros miembros afectados. Los padres no presentan cosanguinidad. Tiene un hermano mayor no afectado del síndrome. En la familia paterna no hay casos.
Las fuentes de información fueron conexión telefónica con la familia directa de la recién nacida, entrevista con su madre, a su vez afectada del síndrome y los archivos del Complejo Asistencial de León.
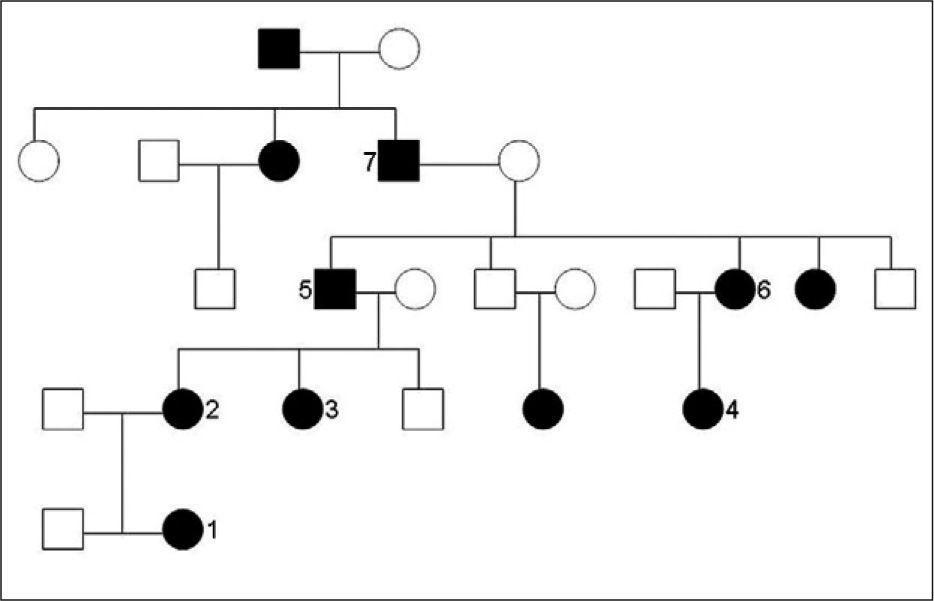
Tras la búsqueda, se encontró que al menos otros 10 miembros de la familia se encuentran afectados, documentados hasta la quinta generación (fig. 1). Contamos con el historial médico de siete de los parientes afectados; en todos ellos apareció el diagnóstico de disostosis cleidocraneal (tabla 1).
Características clínicas de los parientes*
| Fontanelassuturas | Clavículas (Rx de tórax) | Pelvis (Rx de pelvis -caderas) | Cráneo (Rx-TC) | Dismorfia facial | Dentición | Hipoacusia | Partos | Otros | |
| 1 | Bregma amplia. Dehiscencia sutura sagital | Ausencia. Tórax acampanado | Occipucio prominente | Test de audición normal | Cariotipo: normal | ||||
| 2 | Bregma amplia (3 × 3cm). Cierre tardío | Ausencia. Tórax acampanado | Luxación congénita de cadera. Diástasis pubiana | Hipoplasia celdas mastioneas. CAE corto. Huesos wormianos | Hipertelorismo. Prognatismo | Primera retrasada y extraída. Definitiva malformada | Sí. Bilateral | Dos cesáreas por desproporción pelvicocefálica | Retraso pondoestatural. Esguince inveterado de tarso |
| 3 | Bregma amplia. Dehiscencia de sutura sagital. Cieñe tardío. Braquicefalia | Ausencia derecha. Esbozo de izquierda (1/3 interno). Tórax acampanado | Diástasis pubiana | Hipoplasia maxilar inferior. Huesos wormianos | Vitíligo zosteriforme. Quiste ovárico derecho | ||||
| 4 | Bregma “enorme”. Suturas soldadas tardíamente | Ausencia. Tórax acampanado | Hipertelorismo | Múltiples inclusiones dentarias. Exodoncia | Depresión respiratoria en parto | ||||
| 5 | Ausencia de extremos distales | Deformidad | Hipoplasia del maxilar superior | Sí. Bilateral. Porootitis crónica | Prótesis y rescate cadera izquierda. Artrosis de la rodilla izquierda. Criptorquidia derecha | ||||
| 6 | Ausencia | Diástasis. Luxación congénita derecha | Huesos wormianos a nivel occipital | Hipertelorismo. Prognatismo. Epicanto | Exodoncia | Sí. Bilateral | Cesárea por desproporción pelvicocefálica | Coxa vara. Sinusitis crónica. Urticaria-vasculitis | |
| 7 | Parálisis facial izquierda |
CAE: conducto auditivo externo; Rx: radiografía.
Todos los miembros afectados, salvo tres, viven en la actualidad y desarrollan una vida normal.
DISCUSIÓNYa descrita en 1898 por Marie y Saiton5, la disostosis cleidocraneal es una entidad poco frecuente en la que se observa agrupación familiar por su patrón de herencia autosómico dominante1, aunque se ha descrito casos esporádicos. La mutación más frecuente se encuentra en el gen CBFA1/RUNX2, localizado en el cromosoma 6p216–11. Existe una correlación entre el fenotipo y el genotipo8,12.
Se define por la agrupación de una serie de anomalías1–5. La más frecuente es la agenesia o disgenesia de clavículas. A ella se asocian anomalías craneales, como hipoplasia del maxilar superior y fontanela de gran tamaño, facies caracterizada por hipertelorismo, seudoprognatismo y epicanto, problemas dentales e hipoacusia. Puede asociarse la presencia de forámenes en huesos parietales8, otitis media de repetición10 o alteración de la cadera, por la cual es frecuente la realización de cesárea. Las alteraciones de las clavículas permiten una facilidad anormal para oponer y juntar los hombros y, asimismo, la enfermedad se caracteriza por sínfisis del pubis amplia, falanges medias de los quintos dedos de las manos cortas y, a menudo, malformaciones vertebrales. Las características clínicas de los miembros de la familia con cuyo historial contamos se resumen en la tabla 1.
Es posible realizar un diagnóstico prenatal mediante ecografía en embarazadas afectadas13,14. Ante la sospecha clínica de disostosis cleidocraneal en un recién nacido, es conveniente estudiar la presencia de esta entidad en otros miembros de la familia, debido a su patrón de herencia autosómico dominante con penetrancia completa y expresividad variable.
Agenesia o disgenesia clavicularEs característica la formación incompleta o ausencia de clavículas en los sujetos afectados por este síndrome. Se describe y confirma mediante radiografía de tórax en todos los pacientes. Uno presenta un esbozo de la clavícula izquierda y ausencia de la derecha, otro carece de extremos distales de ambas clavículas y el resto presenta ausencia completa de ambas clavículas (fig. 2).
Tórax acampanado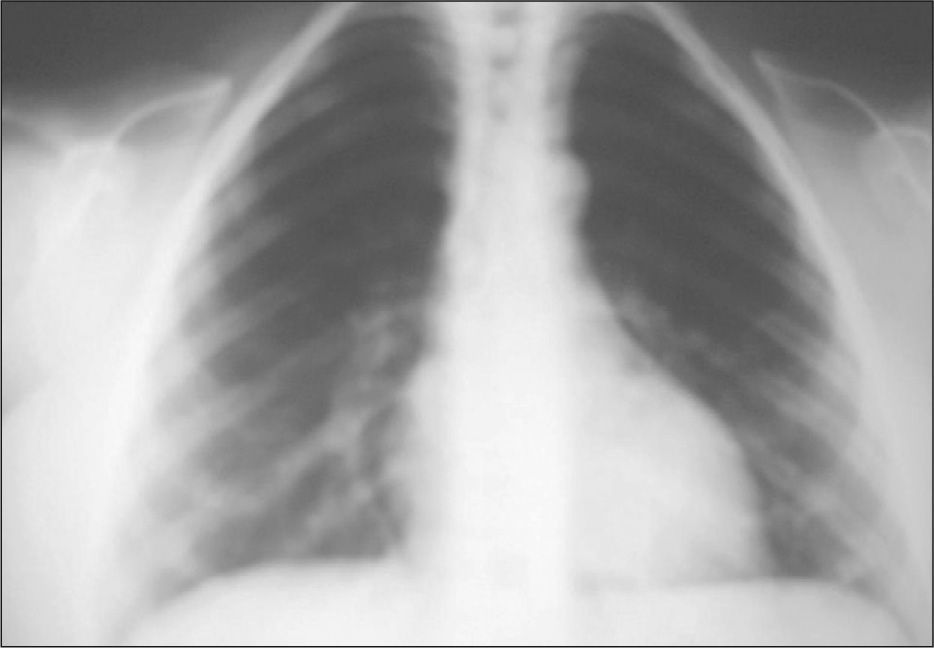
Aparece en los recién nacidos. En los pacientes se solicitó siempre radiografía de tórax, y se observó dicha anomalía en todos ellos.
CráneoEn el cráneo, al nacer, se observan suturas y fontanelas amplias, cuerpo del esfenoides ensanchado y aplanado y presencia de huesos wormianos. En los parientes se describe fontanela bregma de gran tamaño y dehiscencia de la sutura sagital. También se presenta cierre tardío de suturas y fontanelas, confirmado en dos de los miembros de la familia.
En la edad adulta es característica la presencia de hipertelorismo, aplanamiento de la raíz de la nariz, hipoplasia del maxilar superior, prognatismo y persistencia de la sutura mentoniana. Se describen anomalías craneales en cinco de los pacientes. En todos ellos contamos con radiografía de cráneo (fig. 3). En el recién nacido se observa occipucio prominente y afectación del clivus y silla turca. En el resto se aprecia presencia de huesos wormianos e hipoplasia del maxilar superior.
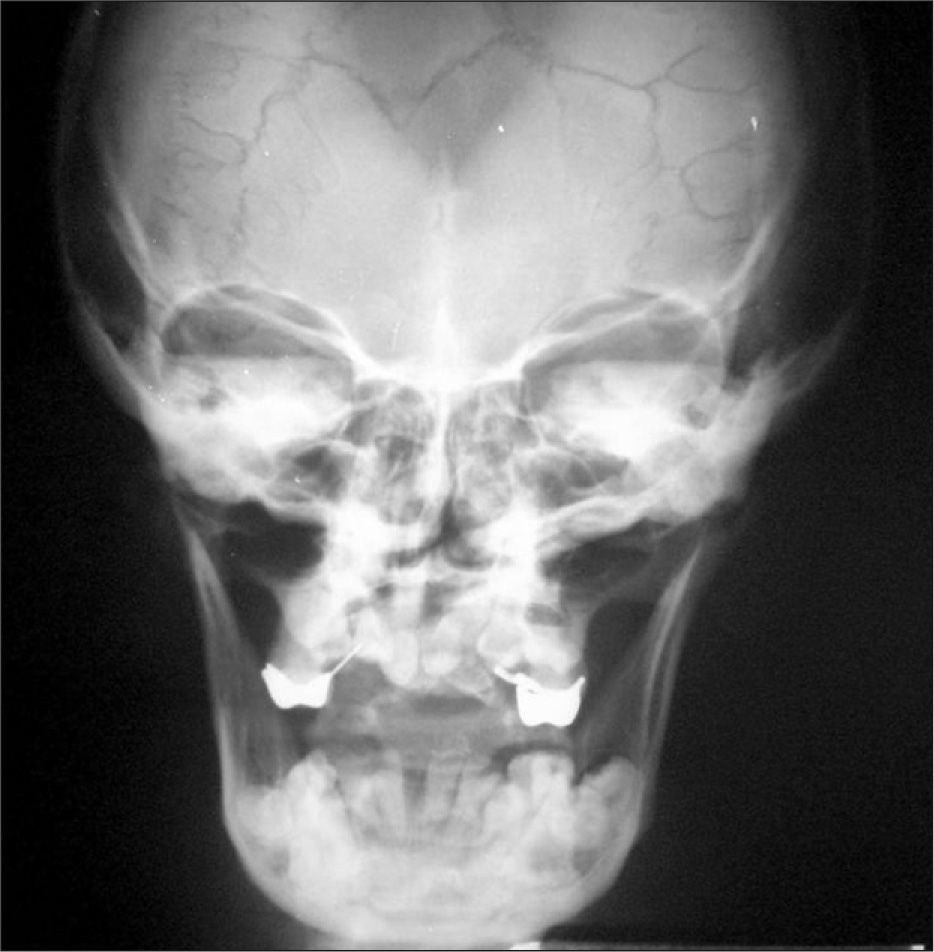
En uno de los casos en que se realizó tomografía computarizada craneal en el contexto de un estudio por hipoacusia se demostró el pequeño desarrollo y neumatización de celdas mastoideas (más acusado en el lado derecho), conductos auditivos externos estrechos (más en el lado izquierdo) e imágenes lineales con contornos de esclerosis a lo largo de ambos peñascos, compatibles con huesos wormianos, así como en el diploe.
En cuatro de los pacientes se describe la facies característica; a continuación se mencionan los siguientes datos en cada uno de ellos: hipertelorismo y prognatismo, hipertelorismo, hipoplasia de maxilar superior, hipertelorismo con prognatismo y epicanto.
La primera dentición aparece de forma tardía, lo cual es normal. La dentición definitiva también es tardía y presenta anomalías de número, localización y esmalte. En una paciente se produjoretraso en la salida de la primera dentición y, posteriormente, retraso en su caída, por lo que se requirió su extracción. Actualmente presenta una dentición definitiva malformada, tributaria de exodoncia. Otros dos fueron intervenidos quirúrgicamente para realizar exodoncia, debido a una malformación y a la presencia de múltiples inclusiones dentarias.
HipoacusiaUna complicación que puede surgir es la hipoacusia. Se debe a las malformaciones de los huesos del cráneo. En tres de los casos se presentan problemas de hipoacusia, en todos ellos bilateral. Uno de los sujetos precisó implantes cocleares bilaterales.
CaderaA la altura de la pelvis existe retraso o ausencia en la osificación de las ramas del pubis e hipoplasia de las alas ilíacas. Dichas alteraciones propician que la mayoría de las mujeres afectadas precisen cesárea en sus embarazos para evitar complicaciones obstétricas.
Se realizó radiografía de cadera a tres de los parientes, y en todos ellos se apreció diástasis de la sínfisis pubiana. Dos de ellos además presentan luxación congénita de la cadera. Los tres partos documentados fueron cesáreas por desproporción pelvicocefálica; los recién nacidos presentaron un peso y un perímetro cefálico normales.
Enfermedades asociadasEn un varón encontramos artrosis precoz de cadera tributaria de prótesis y rescate de la misma (37 años) y artrosis precoz de rodilla. Una mujer presentó retraso pondoestatural y perímetro cefálico conservado en sus primeros años de vida. Otras dos mujeres tuvieron vitíligo zosteriforme y coxa vara, respectivamente.
En resumen, podemos afirmar que hemos llegado a las siguientes conclusiones: 1) el diagnóstico del síndrome se realiza según hallazgos clínicos y radiológicos; 2) las alteraciones tributarias de tratamiento son la hipoacusia, los problemas dentales y la artrosis, y 3) ante el diagnóstico del síndrome es importante realizar un estudio de la familia3.
Presentado en el XIX Memorial Guillermo Arce y Ernesto Sánchez Villares. Salamanca, 20 y 21 de octubre de 2006.
