
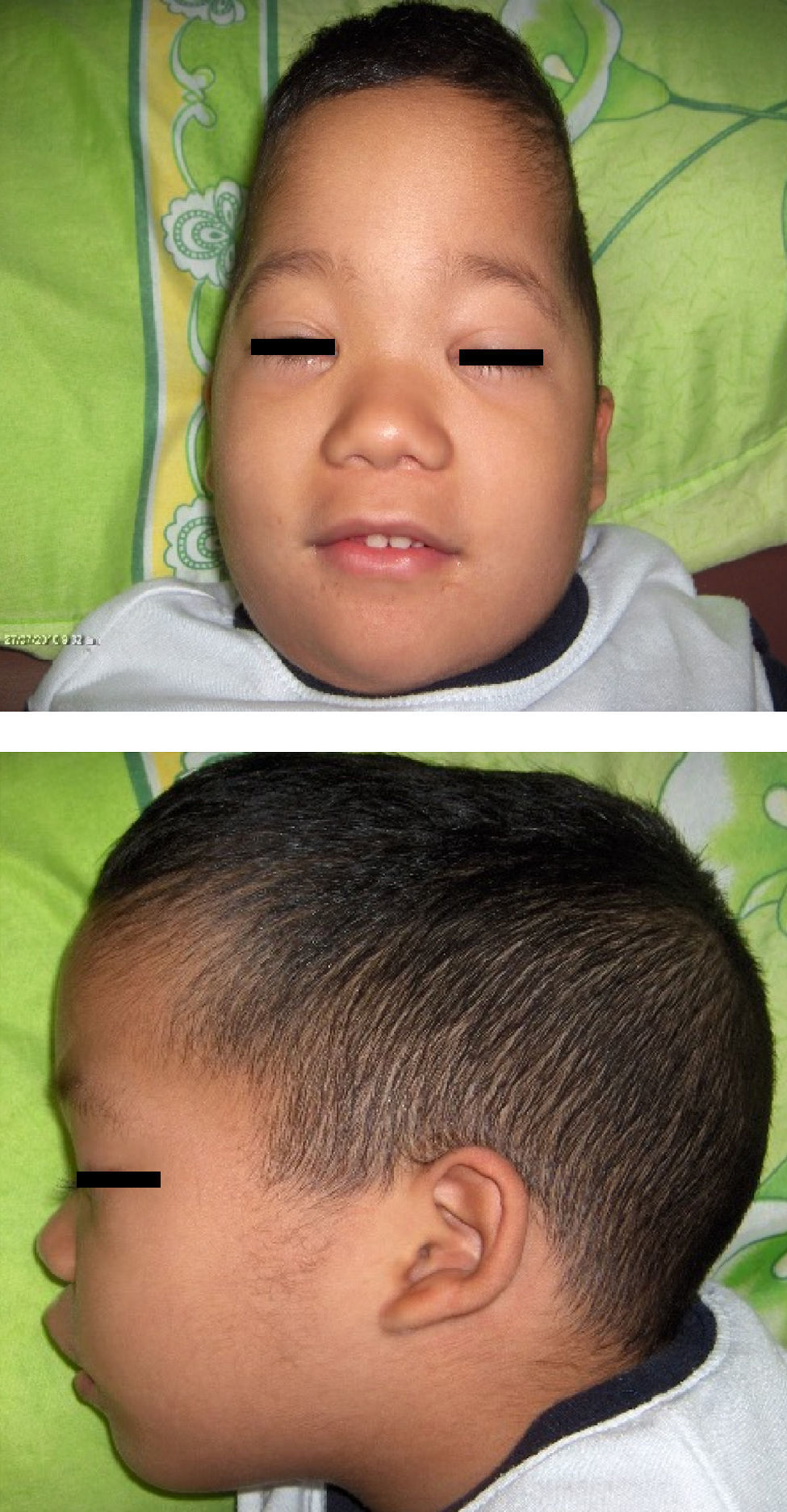
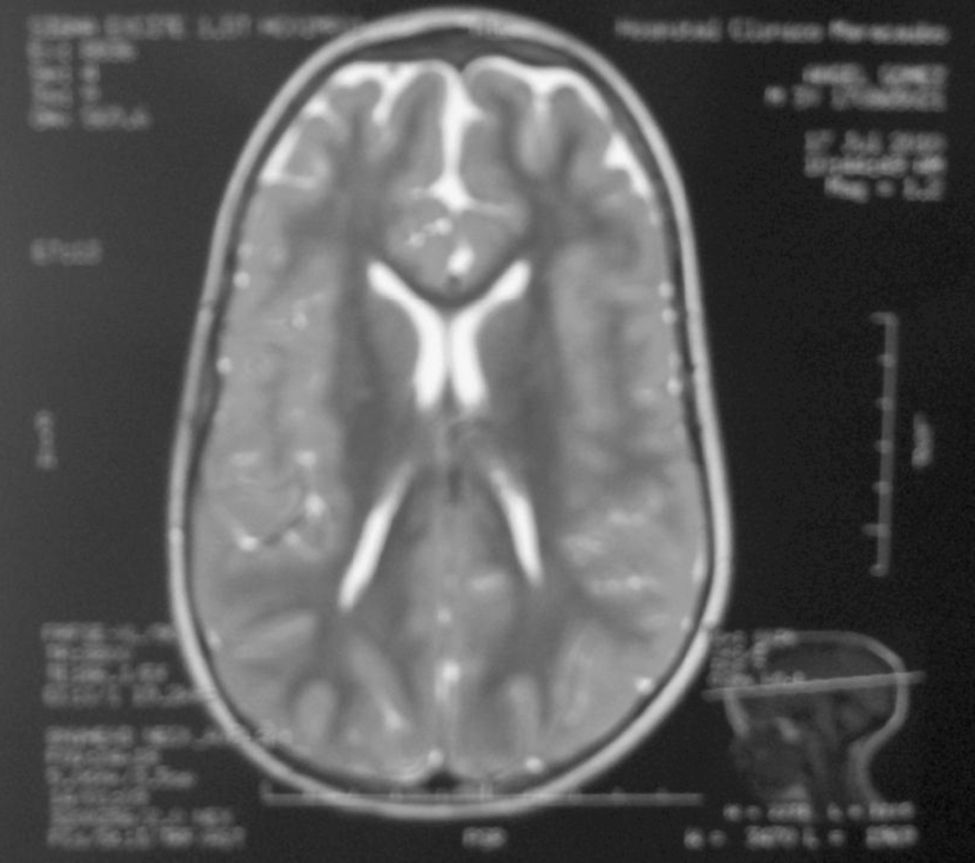
La aciduria cetoglutárica ha sido asociada con enfermedad neurológica con retraso del desarrollo psicomotor, convulsiones, ventriculomegalia, atrofia de corteza cerebral frontal y biparietal anterior, cisura de Silvio amplia, pero no con dismorfias faciales. Presentamos un paciente masculino con diagnóstico bioquímico de aciduria cetoglutárica por la presencia de hiperlacticinemia y excreción urinaria de 2-cetoglutárico en dos oportunidades. Igualmente presentaba dismorfias faciales no reportadas como dolicocefalia, frente prominente, fontanela anterior abierta, epicanto bilateral, nariz pequeña, puente nasal ancho y deprimido, columela corta, filtrum largo y pabellones auriculares dismórficos de implantación baja (fig. 1). Además, presentó criptorquidia derecha, hipotonía troncal, convulsiones, ventriculomegalia, atrofia de corteza cerebral frontal y biparietal anterior, cisuras de Silvio amplias (fig. 2); Cariotipo en sangre periférica (950 bandas) 46,XY y hormonas tiroideas normales. Las manifestaciones neurológicas, neuroimágenes y bioquímicas sugieren el diagnóstico aciduria cetoglutárica debido a deficiencia del complejo 2-α-cetoglutarato deshidrogenasa. Las dismorfias faciales descritas no han sido reportadas previamente.


El complejo de la 2-α-cetoglutarato deshidrogenasa pertenece al grupo de los complejos de α-cetoácidos deshidrogenasas y se ubica en el metabolismo del ciclo de Krebs; está constituido por tres subunidades polipeptídicas: oxoglutarato deshidrogenasa (E1k), dihidrolipoil succiniltransferasa (E2k) y dihidrolipoil deshidrogenasa (E3). La deficiencia de alguno de sus componentes bloquea la actividad enzimática del complejo en el ciclo de Krebs con disminución en la producción de energía y acumulación de componentes tóxicos y en consecuencia desarrollo de enfermedad neurológica. Las manifestaciones neurológicas asociadas son ampliamente variables: retardo mental y convulsiones1, opistótonos, hipertonía troncal, hiperexcitabilidad2, hipotonía axial, comportamiento sicótico y síntomas piramidales3.
Las dismorfias craneo-faciales observadas, tales como dolicocefalia, frente prominente, fontanela anterior abierta, epicanto bilateral, nariz corta, puente nasal ancho y deprimido, columela corta, filtrum largo y pabellones auriculares dismórficos y de baja implantación, no han sido reportadas en la aciduria α-cetoglutárica por deficiencia enzimática del complejo de la α-cetoglutárica deshidrogenasa; éste es el primer caso que se publica, pero son similares a las reportadas en la deficiencia del complejo de la piruvato deshidrogenasa4–8, donde el 35% de los pacientes pueden presentar estas dismorfias faciales4–6.
Los diagnósticos diferenciales planteados son las deficiencias de los complejos de la piruvato deshidrogenasa, el complejo de la α-cetoácidos de cadena ramificada deshidrogenasa o la deficiencia combinada de los complejos de α-cetoácidos deshidrogenasas, ya que la dihidrolipoamida deshidrogenasa (E3) es común a los tres complejos. La deficiencia combinada de estos complejos es infrecuente9 y se detectan excreción de lactato, piruvato, α-hidroxibutirato, α-hidroxiisovalerato y α-cetoglutarato10. De la misma manera, nuestro paciente no mostró excreción urinaria de metabolitos por deficiencia del complejo de la α-cetoácidos de cadena ramificada deshidrogenasa. Excluyendo la posibilidad anterior, la excreción urinaria α-cetoglutarato descarta la deficiencia del complejo de la piruvato deshidrogenasa aun cuando se evidenció atrofia cerebral, ventriculomegalia y ampliación de las cisuras de Silvio pero no lesiones en los ganglios de la base. La ampliación de las cisuras de Silvio puede sugerir el diagnóstico de aciduria glutárica I o aciduria glutárica II, pero la ausencia de metabolitos marcadores de 2-OH-glutárico y 3-OH-glutárico las descarta.
En conclusión, hasta la fecha no se reportan dismorfias faciales asociadas a la aciduria α-cetoglutárica por déficit del complejo de la α-cetoglutárica deshidrogenada, las cuales contribuirán a la delineación fenotípica del síndrome.

