



Introducción
La clasificación nosológica de los pacientes con afectación congénita de múltiples pares craneales, en nuestra opinión, no está adecuadamente resuelta. A los recién nacidos o lactantes en los que predomina la parálisis de la musculatura facial y/o oculomotora se les describe bajo el nombre de síndrome de Möbius o secuencia de Möbius. A los recién nacidos o lactantes con retromicrognatia, dificultad respiratoria y disfunción deglutoria, con o sin paladar hendido, se les suele encontrar descritos bajo el nombre de síndrome de Pierre Robin o secuencia Pierre Robin. Este tipo de clasificación, práctica por la facilidad para recordar los epónimos y por su amplio uso a lo largo de los años, no se ciñe a la realidad, ya que es difícil encontrar 2 pacientes con idénticas manifestaciones clínicas, muchos de ellos presentan hipotonía y otros signos de afectación del tronco del encéfalo y son pocos los pacientes que cumplen con los criterios restringidos del síndrome de Möbius propuestos por varios autores1,2 o los propuestos por Jones, en 19873, para el síndrome de Pierre Robin.
Etiología y patogénesis
Para explicar el origen de los síndromes de Möbius4, Pierre Robin5 y Cogan6 se han propuesto numerosas causas y varias teorías patogénicas que implican tanto procesos genéticamente determinados como alteraciones adquiridas durante el período gestacional, pero ninguna de ellas justifica la variabilidad de la sintomatología ni la disparidad de los hallazgos anatomopatológicos2,3,7. Se han descrito pacientes con agenesia de núcleos motores, ausencia de la porción nerviosa y/o hipoplasia de los músculos inervados por pares craneales motores. Un considerable número de pacientes, la mayoría de ellos publicados como casos de síndrome o secuencia de Möbius eran esporádicos y, aparte de los hallazgos propios del síndrome, presentaron hipotonía muscular y trastornos de la deglución. Además, en algunos de ellos se observaron varias combinaciones: trismo, disfunción respiratoria central y reflujo gastroesofágico. Muchos de ellos fallecieron durante los primeros meses de vida y en aquellos en los que se pudo practicar estudios post mortem se observaron lesiones necróticas antiguas en las zonas paramediales del tegmento que se extendían desde el mesencéfalo hasta la porción superior de la médula oblongada8-15. Aun más, Hagerman et al16, en su estudio clinicopatológico de 65 pacientes con hipocinesia fetal encontraron lesiones del tronco del encéfalo en los 2 pacientes afectados de síndrome de Möbius y que también presentaban contracturas articulares congénitas.
La patogenia de los diferentes síndromes que nosotros incluimos bajo la denominación genérica de disgenesia troncoencefálica, empieza a ser comprendida. Desde la vertiente clínica, el estudio realizado en Holanda por Verzijl et al17, en una serie de 37 sujetos catalogados como síndrome de Möbius, demuestra que las manifestaciones clínicas de estos pacientes son mucho más amplias que las descritas inicialmente por este autor y sólo pueden explicarse si se entiende al síndrome de Möbius como síndrome de "mal desarrollo" del romboencéfalo.
La apraxia oculomotora congénita o síndrome de Cogan se ha descrito de forma aislada o asociada a varias anomalías del sistema nervioso central (SNC), así como en algunas enfermedades neurodegenerativas18. Más de la mitad de los pacientes con síndrome de Cogan "aislado" sufren trastornos oromotores, hipotonía y retraso del desarrollo19. La causa o causas y la localización anatómica que provocan la apraxia oculomotora congénita no se conocen con certeza. Se han implicado los lóbulos frontales, cerebelo y la porción superior del tronco del encéfalo como posibles lugares de origen del síndrome de la apraxia oculomotora congénita. Cogan6, en su descripción inicial, propuso que el síndrome se debía a una detención del desarrollo o de la mielinización de las vías involucradas en el control de la mirada conjugada. En el síndrome descrito por nosotros20 la lesión que causa la apraxia oculomotora que presentan los pacientes es sin duda troncoencefálica, y probablemente protuberancial, en el centro que controla los movimientos sacádicos horizontales.
En los estudios llevados a cabo en los últimos 10 años por Abadie et al21 en un conjunto de 66 pacientes diagnosticados de secuencia de Pierre Robin se concluye que la sintomatología de estos pacientes no puede explicarse únicamente sobre la base de las alteraciones anatómicas óseas y, al igual que otros autores, proponen que la causa de las manifestaciones clínicas de este tipo de pacientes tiene una patogenia neuroembriológica que da lugar a disfunción de las estructuras troncoencefálicas durante el desarrollo fetal.
El grupo de pacientes descrito por nosotros representa probablemente el síndrome más grave de disgenesia troncoencefálica compatible con la supervivencia posnatal. Uno de los pacientes de nuestra serie falleció a consecuencia de una aspiración alimentaria masiva. En el estudio neuropatológico macroscópico no se apreciaron anomalías excepto una ligera atrofia del tronco del encéfalo. El examen microscópico no puso de manifiesto alteraciones en la corteza cerebral, amígdalas, sustancia gris central y cerebelo. La sustancia negra, colículos, núcleo rojo, locus caeruleus, núcleos pontinos, olivas inferiores y núcleo ambiguo estaban así mismo conservados; sin embargo, se apreció pérdida neuronal y gliosis en los núcleos de los pares craneales IV, VII, VIII y IX, junto con gliosis en la formación reticular medial de la médula oblongada. La naturaleza de estas lesiones se consideró de tipo destructivo/disruptivo por la presencia de gliosis difusa y por la afectación de tractos corticales descendentes (20). Los estudios patológicos experimentales de Leong y Ashwell22, por otra parte, demuestran que las zonas mediales y paramediales del tegmento del tronco del encéfalo durante el desarrollo embrionario están poco vascularizadas en relación con las estructuras localizadas más lateralmente. Finalmente, Sarnat, en un artículo reciente23, propone que infartos en las zonas limítrofes (watershed infarcts) del tronco del encéfalo durante el período embrionario o fetal son los responsables de la mayoría de síndromes que cursan con afectación congénita múltiple de pares craneales.
Clasificación de la disgenesia troncoencefálica
El término disgenesia deriva de la combinación de los vocablos griegos dys y gennan y se utiliza en muchos campos de la medicina para indicar el desarrollo defectuoso, parcial o total, de un órgano. Existe controversia con relación al uso del término disgenesia. Algunos autores opinan que debe restringirse a las alteraciones que se producen durante el período embrionario. Estos autores quieren dotar al término disgenesia de una connotación estrictamente genética-hereditaria. Otros autores aceptan que la palabra disgenesia puede aplicarse al desarrollo defectuoso a lo largo de toda la gestación y, en consecuencia, aceptan que las anomalías pueden ser debidas tanto a causas determinadas genéticamente como a procesos patológicos que se produzcan durante la gestación. Nosotros preferimos esta acepción más amplia ya que es difícil, basándose en las manifestaciones clínicas, determinar el momento de la lesión y, por otra parte, se han podido documentar lesiones vasculares del tronco del encéfalo durante los primeros meses de la gestación22,24.
De acuerdo con nuestro enfoque la disgenesia troncoencefálica puede clasificarse (tabla 1) en genéticamente determinada1,25-27 y adquirida y ambas, a su vez, pueden estar asociadas o no a otras malformaciones. Las formas asociadas a otras malformaciones probablemente comparten mecanismos de producción de las anomalías formal genesis syndromes28 y algunos autores las agrupan bajo el nombre de oro-mandibular limb hypogenesis syndromes7 mientras que otros prefieren la denominación terminal transverse defects with orofacial malformations28. Las manifestaciones clínicas de los casos adquiridos destructivos o disruptivos, al igual que sucede con las lesiones vasculares de los hemisferios cerebrales, dependerán del territorio vascular involucrado y de la extensión del parénquima afectado (tablas 2 y 3). La mayoría de los pacientes descritos bajo los nombres de síndromes y secuencias de Möbius, Pierre Robin y Cogan, se deben a alteraciones disruptivas del tronco del encéfalo durante el período de gestación y, en consecuencia, deben ser entendidos como formas de presentación de disgenesia troncoencefálica. Los epónimos deberían utilizarse tan sólo en aquellos pacientes que cumplieran con los hallazgos descritos inicialmente por cada uno de los citados autores.

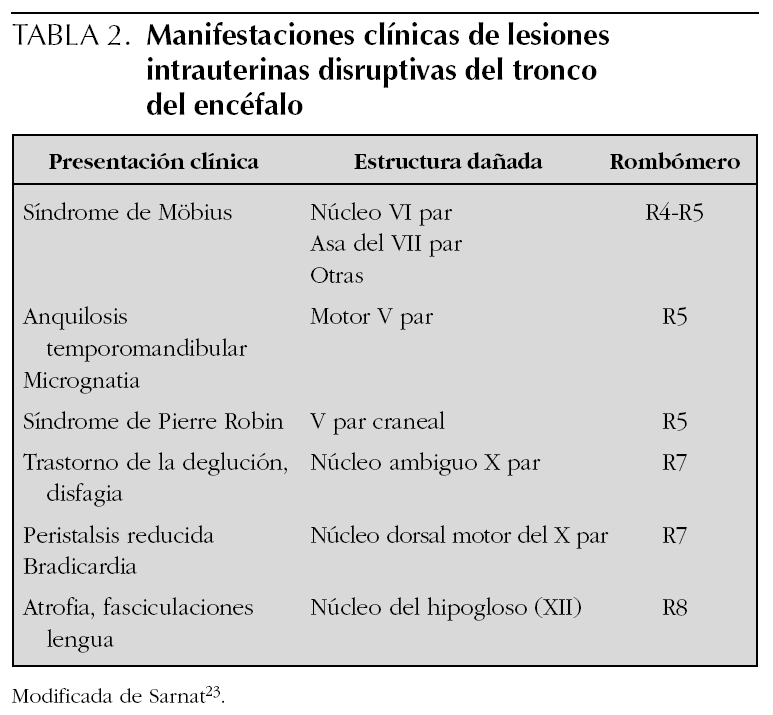
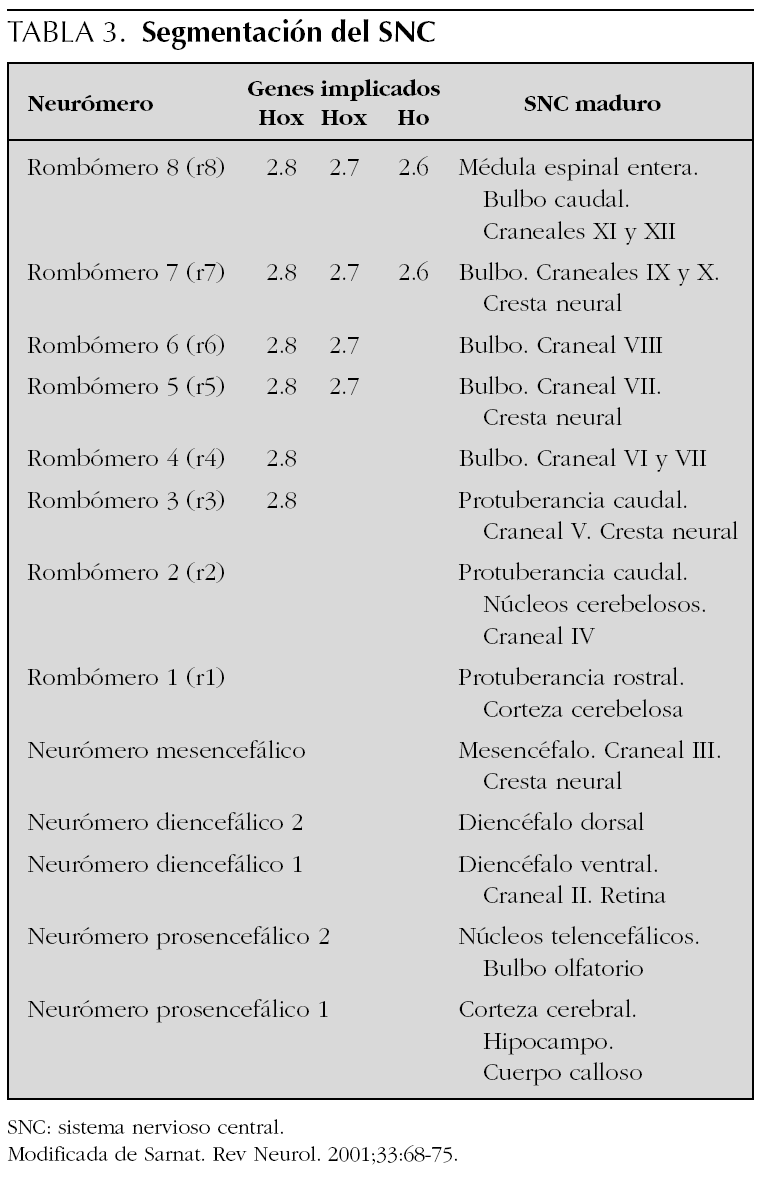
Estudios diagnósticos
El diagnóstico de disgenesia troncoencefálica en recién nacidos con afectación congénita de múltiples pares craneales puede ser difícil de establecer a no ser que se sospeche y que, desde el principio, se estudie a estos niños minuciosamente. Debemos hacer notar que en muchos casos la sintomatología mejora con el paso del tiempo20 y la información clínica puede perderse o interpretarse incorrectamente en análisis retrospectivos. No es infrecuente encontrar signos piramidales en pacientes con disgenesia troncoencefálica y por lo tanto es posible que alguno de ellos se incluyan dentro de las formas atípicas de parálisis cerebral29 y, en consecuencia, reciban un peor pronóstico del que les corresponde.
Los estudios neurorradiológicos en muchos casos de disgenesia troncoencefálica son poco informativos. Ocasionalmente, la tomografía computarizada puede detectar calcificaciones troncoencefálicas11,12,15. En algunos pacientes, especialmente adultos, la resonancia magnética (RM) ha puesto de manifiesto anomalías del tronco del encéfalo que apoyan el diagnóstico de síndrome de Möbius30 o el de Cogan31. No obstante, el examen del SNC de estos pacientes utilizando la RM es indispensable, ya que permite excluir otros procesos patológicos asociados, o definir mejor aquellos pacientes con trastornos oromotores congénitos asociados a malformaciones del SNC, sobre todo el síndrome biopercular o perisilviano29,32 con los que se puede confundir. La radiología convencional puede ser de gran ayuda en aquellos casos en los que pone de manifiesto la presencia de malformaciones óseas, anquilosis temporomandibular, incoordinación velopalatina o reflujo gastroesofágico.
Los diversos estudios neurofisiológicos de los que se dispone en la actualidad (velocidad de conducción nerviosa, electromiografía, estimulación motora repetitiva, blink reflex, potenciales evocados de tronco, potenciales somestésicos, electroencefalograma nocturno con polisomnografía) son difíciles de practicar e interpretar en recién nacidos y lactantes y, sin embargo, son de enorme importancia para poder confirmar la localización y la extensión de la lesión en los pacientes con disgenesia troncoencefálica. En nuestra opinión y la de otros autores20,33,34 el uso combinado de los diferentes estudios neurofisiológicos disponibles es el procedimiento que proporciona mayor información diagnóstica. En la mayoría de los pacientes con disgenesia troncoencefálica estudiados por nosotros hemos encontrado, en diferentes combinaciones, alteraciones de alguno de los componentes del blink reflex, patrones neurógenos en alguno de los músculos faciales, latencias III-V alargadas en los potenciales de tronco y alteraciones durante el sueño REM en los estudios polisomnográficos. Algunos autores, de forma experimental, están llevando a cabo estudios neurofisiológicos muy interesantes sobre el proceso normal y patológico de la succión y de la deglución en recién nacidos35 que en el futuro complementarán el acervo diagnóstico de la disgenesia troncoencefálica.
La hipotonía, especialmente durante los primeros meses de la vida, puede ser un hallazgo clínico tan llamativo como la afectación de los pares craneales, hasta el punto que puede inducir a la sospecha de enfermedad neuromuscular. En estos casos, estudios electromiográficos, metabólicos, incluyendo la biopsia muscular, pueden estar indicados para excluir o confirmar esta hipótesis diagnóstica. En los pacientes de nuestra serie20 la hipotonía muscular era muy evidente. Este hallazgo, combinado con un electromiograma de características miógenas, obligó a practicar en uno de los pacientes una biopsia muscular que resultó normal.
Algunos pacientes, especialmente los que presentan lesiones localizadas en la porción caudal del tronco del encéfalo, padecen alteraciones graves del ritmo cardíaco que pueden ocasionar la muerte del paciente. Las arritmias, que a veces se identifican durante la práctica del electroencefalograma convencional o de la polisomnografía nocturna, deben confirmarse o descartarse con estudios prolongados del ritmo cardíaco21.
Por último, no hay que olvidar que la disgenesia troncoencefálica también puede ser debida a alteraciones genéticamente determinadas. Estudios experimentales han puesto de manifiesto que anomalías en genes organizadores o reguladores pueden ser la causa de malformaciones de la línea media facial, del tórax y de estructuras troncoencefálicas36-39. En los últimos años hemos podido observar cómo deleciones de la región q11.2 del cromosoma 22 pueden ser responsables de varios síndromes, dentro de los cuales podrían verse incluidas formas leves de disgenesia troncoencefálica40-42). En consecuencia, es recomendable descartar anomalías de la región 22q11.2 en todos aquellos pacientes con sospecha diagnóstica en los que predominan los trastornos de succión-deglución y, especialmente, los asociados a malformaciones cardíacas. La forma neonatal de la distrofia miotónica y las formas precoces de atrofia muscular espinal infantil pueden confundirse con la disgenesia troncoencefálica, por lo que, dada la transmisión genética y la gravedad de las mismas, deberían practicarse estudios diagnósticos moleculares de ambas enfermedades, ante la más mínima duda diagnóstica.
Pronóstico y tratamiento
El pronóstico y tratamiento de estos pacientes depende del tipo de afectación y, en el caso de tratarse de procesos destructivos/disruptivos del tronco del encéfalo, de la magnitud de éstos. Es importante resaltar, sin embargo, que algunos recién nacidos pueden presentar una afectación troncoencefálica reversible que se ha descrito bajo los nombres: Transient pharyngeal incoordination in the newborn43, retard de maturation de la succion-déglutition44 o dysfonctionnement néonatale isolé du tronc cerebral45 cuyo pronóstico, al tratarse probablemente de un proceso dismadurativo, es excelente.
Las mayores secuelas y el pronóstico vital de los pacientes con disgenesia troncoencefálica vienen determinados por la intensidad del trastorno de deglución y la importancia del reflujo gastroesofágico, a los que pueden sumarse alteraciones del ritmo cardíaco. En algunos pacientes se hace precisa la alimentación por gastrostomía y, en estos casos, es muy importante verificar el grado de reflujo gastroesofágico concomitante. En los lactantes con reflujo grave es preciso el tratamiento quirúrgico paliativo para evitar aspiraciones alimentarias graves. En cualquier caso, las aspiraciones repetidas de alimentos pueden ser la causa de ingresos múltiples y de lesiones pulmonares crónicas.
El pronóstico funcional de los pacientes con disgenesia troncoencefálica es mucho más difícil de establecer ya que, según los niveles del tronco involucrados, pueden verse afectadas distintas estructuras. Los pacientes con diplejía facial, aparte de las dificultades iniciales para la succión o la deglución, presentarán pérdida de la mímica facial, la consiguiente dificultad de comunicación y las repercusiones psicológicas que pueden derivar de ésta. La incoordinación velopalatina, la diplejía facial y la anquilosis temporomandibular, de nuevo, aparte de las dificultades iniciales para la alimentación, posteriormente son la causa de las alteraciones de la articulación del lenguaje que con frecuencia presentan este tipo de pacientes. Las anomalías de la ventilación tubárica, muy comunes en estos niños, predisponen a la otitis serosa de repetición y a las complicaciones derivadas de ésta.
Finalmente, hay que recordar que en la mayoría de pacientes que presentan disgenesia troncoencefálica, esta se debe a procesos disgenéticos destructivos o disruptivos que ocurren durante los primeros meses de la gestación. La naturaleza de estos procesos disgenéticos es esporádica y no comporta riesgo de recurrencia familiar20,23.
