



La asociación de ictericia neonatal prolongada e hipoglucemia recurrente puede ser secundaria a una patología endocrinológica subyacente. La insuficiencia hipofisaria y la insuficiencia adrenal primaria son las principales patologías que se deben descartar.
Material y métodosSe analizaron retrospectivamente las características clínicas y de laboratorio de 13 pacientes derivados a la división de endocrinología del Hospital de Niños Ricardo Gutiérrez entre los años 2003 y 2008 con ictericia neonatal e hipoglucemia secundaria a insuficiencia hipofisaria en 12 pacientes y en uno a insuficiencia adrenal primaria.
ResultadosTodos los pacientes tuvieron hipoglucemia en el periodo neonatal. En 10 pacientes la hiperbilirrubinemia fue de predominio directo y 6 pacientes presentaron elevación de transaminasas. La insuficiencia hipofisaria fue múltiple en los 12 pacientes. El tratamiento de remplazo hormonal normalizó la función hepática, resolvió la ictericia en todos los niños y ninguno requirió biopsia hepática. Los episodios de hipoglucemia también cedieron al iniciar el tratamiento sustitutivo.
ConclusionesEl binomio ictericia prolongada o colestásica e hipoglucemia recurrente exige descartar insuficiencia hipofisaria múltiple e insuficiencia suprarrenal primaria. La terapia sustitutiva correspondiente resuelve el problema colestásico en la mayor parte de los casos, así como los problemas derivados de la hipoglucemia recurrente y las deficiencias hormonales.
The association of prolonged neonatal jaundice and hypoglycaemia may be secondary to an endocrinological disease. Pituitary insufficiency and primary adrenal insufficiency are the most likely endocrine diseases that need to be ruled out.
Material and methodsWe retrospectively analysed the clinical and laboratory characteristics of thirteen patients referred to the Hospital de Niños Ricardo Gutiérrez between years 2003 and 2008 due to prolonged neonatal jaundice and hypoglycaemia secondary to pituitary insufficiency in twelve patients, and in one secondary to primary adrenal insufficiency.
ResultsAll patients had a history of neonatal hypoglycaemia. Ten patients had conjugated hyperbilirubinaemia and six also had elevated transaminases. Combined pituitary hormone deficiency was observed in the twelve hypopituitarism patients. Hormonal replacement normalised liver function and resolved the prolonged jaundice in all the patients. None of them underwent liver biopsy. Hypoglycaemia also remitted after hormonal therapy.
ConclusionsProlonged or cholestatic jaundice associated with neonatal hypoglycaemia is highly likely to be due to pituitary hormone deficiency or primary adrenal insufficiency. Early diagnosis and treatment of these children reverts the prolonged jaundice and prevents morbidity and mortality due to recurrent hypoglycaemia and hormone deficiencies.
Endocrinopatías congénitas como la insuficiencia hipofisaria y la insuficiencia adrenal primaria suelen presentar manifestaciones clínicas durante el periodo neonatal y/o durante los primeros meses de vida. Entre ellas se destacan la hipoglucemia y la ictericia prolongada, en particular de tipo colestática. La falta de reconocimiento precoz de estas patologías habitualmente se asocia a una significativa morbilidad y ocasional mortalidad que asciende al 14% según Vulsma et al.1. Es por ello que algunos autores enfatizan la importancia de considerar entre los diagnósticos diferenciales de colestasis neonatal a la insuficiencia hipofisaria (IH) y a la insuficiencia adrenal tanto primaria (IAP) como central, especialmente si se asocian con hipoglucemia2–6. El propósito del presente trabajo fue describir los parámetros clínicos y de laboratorio de una cohorte de niños con IH o IAP, cuya manifestación clínica predominante en los primeros meses de vida fue la ictericia prolongada y la hipoglucemia neonatal.
Pacientes y métodosSe utilizó un diseño retrospectivo y observacional (serie de casos). Se incluyó a 13 niños (9 varones) con diagnóstico de IH o IAP que fueron referidos a la división de endocrinología del Hospital de Niños Ricardo Gutiérrez de Buenos Aires entre los años 2003 y 2008 por presentar ictericia e hipoglucemias desde el periodo neonatal. La edad de consulta (mediana) fue de 34 días de vida (rango 13-94 días).
Se analizaron las historias clínicas de los pacientes, recolectándose los parámetros clínicos, bioquímicos e imagenológicos, tanto en el momento de su admisión como durante la evolución. Como variables clínicas, se recabaron la edad gestacional, el peso de nacimiento, la edad en el momento de la consulta endocrinológica y la presencia de signos clínicos sugestivos de defectos de línea media, anomalías genitales en varones (micropene, criptorquidia y/o microorquidismo) o hiperpigmentación cutaneomucosa.
Dentro de las variables bioquímicas se consignaron las determinaciones de glucemia, bilirrubinemia directa e indirecta, transaminasas glutámico pirúvica (GPT) y glutámico oxalacética (GOT) y gammaglutamiltranspeptidasa (GGT). Se definió hipoglucemia como glucemia plasmática menor a 40mg/dl, hiperbilirrubinemia al nivel de bilirrubina total mayor a 12mg/dl (en menores de 1 mes de edad) y colestasis a la concentración de bilirrubina conjugada mayor al 15-20% de la bilirrubina total durante más de 14 días7.
Se definió IH múltiple a la deficiencia simultánea de 2 o más trofinas hipofisarias. Para el diagnóstico de insuficiencia hormonal se evaluó la función de los distintos ejes hipotálamo hipofisarios y se utilizaron los criterios aplicados en nuestra división:
- –
Insuficiencia de hormona de crecimiento (GH): GH basal en hipoglucemia<10ng/ml o<2ng/ml en normoglucemia durante el periodo neonatal o GH<6ng/ml bajo prueba de estímulo con arginina en niños mayores de un año que evolucionaron con retardo de crecimiento (CLIA, Immulite 2000, Siemens, Alemania), asociado a nivel de IGF-I<-2 DS para la edad (RIA extractivo desarrollado). Hipotiroidismo central: tiroxina libre (T4 l) baja (< 0,8ng/dl) asociado a tirotrofina (TSH) baja, normal, o ligeramente aumentada (ECLIA, Elecsys 2010, Roche Diagnostics GMBH, Mannheim, Alemania). Insuficiencia adrenal: cortisol sérico<1,1μg/dl en menores de 2 meses de edad y<2,1μg/dl en mayores de 2 meses de edad en normoglucemia o<18μg/dl en hipoglucemia (CLIA, Immulite 2000, Siemens, Alemania). La misma se definió central cuando los niveles de ACTH plasmática basal se encontraban normales (10-46pg/ml) o disminuidos y primaria con niveles de ACTH por encima del rango normal (CLIA, Immulite 2000, Siemens, Alemania). Insuficiencia gonadotrófica: LH<0,8 U/l, FSH<0,45 U/l (IFMA, Wallac, Perkin Elmer, Finlandia) y testosterona<10ng/dl (ECLIA, COBAS, Roche Diagnostics GMBH, Mannheim, Alemania) en varones y FSH<1,5 U/l en niñas. La deficiencia de Prolactina (CLIA, Immulite 2000, Siemens, Alemania) fue definida de acuerdo a niveles establecidos de la bibliografía8.
Los estudios por imágenes analizados fueron: ecografía abdominal para evaluación hepática y suprarrenal, gammagrafía de excreción biliar con derivados iminodiacéticos marcados con 99Tc (IDA) para descartar atresia de vías biliares y resonancia magnética (RM) de cerebro con y sin gadolinio para evaluar la región selar y supraselar en los pacientes con IH.
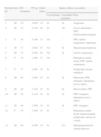
ResultadosDoce niños tenían diagnóstico de IH (del caso 1 al 12) y un niño IAP (caso 13). La tabla 1 muestra los antecedentes perinatales y las características clínicas de los pacientes.
Características clínicas de los pacientes
| Paciente (#) | Sexo | EG (semanas) | PN (g) | Edad (días) | Signos clínicos asociados | ||
| Convulsiones | Anomalías genitales | Otros | |||||
| 1 | M | 38 | 2.950 | 34 | Sí | Sí | Angiomas |
| 2 | M | 34 | 2.520 | 40 | Sí | No | Facies dismórfica, FPP, hepatoesplenomegalia |
| 3 | F | 36 | 3.300 | 13 | No | – | FPP, distrés respiratorio, hipotonía |
| 4 | M | 37 | 2.620 | 87 | No | Sí | Hipotermia/estrabismo |
| 5 | M | 35 | 2.620 | 94 | No | Sí | Distrés respiratorio |
| 6 | F | 34 | 1.890 | 31 | No | – | Hipoplasia medio facial, FPP, distrés respiratorio |
| 7 | F | 37 | 2.980 | 76 | Sí | – | Polidactilia, hernia umbilical |
| 8 | M | 40 | 3.500 | 47 | Sí | Sí | Hipotonía, FPP, nistagmo, hipoplasia nervio óptico |
| 9 | M | 40 | 3.225 | 16 | Sí | Sí | Microcefalia, FPP |
| 10 | M | 39 | 4.120 | 32 | Sí | Sí | FPP, nistagmo, hipoplasia nervio óptico |
| 11 | M | 40 | 3.500 | 14 | Sí | Si | FPP, nistagmo |
| 12 | F | 39 | 3.540 | 53 | Sí | – | Hipoplasia medio facial, hepatomegalia, polidactilia, atresia de coanas |
| 13 | M | 40 | 4.200 | 14 | Sí | No | Hiperpigmentación cutaneomucosa |
EG: edad gestacional; FPP: fontanela posterior permeable; PN: peso al nacer.
Las hipoglucemias se constataron en el periodo neonatal en 12 pacientes, en 5 de ellos durante las primeras 24 h de vida. En un paciente se detectó la hipoglucemia al tercer mes de vida durante el estudio de la colestasis. Sin embargo, la anamnesis dirigida sugirió la presencia de síntomas compatibles con hipoglucemia durante el periodo neonatal (hipotonía e hipotermia). Las hipoglucemias fueron recurrentes en todos los niños y en la mayoría sintomáticas, 9 de ellas graves. Todos los pacientes presentaron concentraciones séricas de insulina adecuadas para el nivel de glucemia.
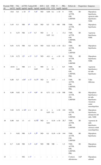
En 10 pacientes la hiperbilirrubinemia fue a predominio directo (colestasis) y en tres a predominio indirecto (casos 1, 3 y 10) (tabla 2). La colestasis neonatal correspondió a IH en nueve pacientes y a IAP en uno. Los 3 pacientes con hiperbilirrubinemia de predominio indirecto presentaron IH. Cinco de los pacientes con colestasis manifestaron también patrón de hepatitis (aumento de las transaminasas de 3 a 4 veces sobre el valor máximo de referencia normal), correspondiendo al paciente con IAP y 4/9 pacientes con IH. El nivel de GGT se evaluó en 6 pacientes con IH; en 4 fue normal y en 2 ligeramente aumentada (entre 1,7 a 2 veces sobre el rango máximo para la edad). En el paciente con IAP se encontró elevada en niveles similares (tabla 2).
Hepatograma y glucemia de los pacientes al diagnóstico
| Paciente (#) | BiT (mg/dl) | BiD (mg/dl) | TGO (U/l) | TGP (U/l) | GGT (U/l) | Glucemia (mg/dl) |
| 1 | 9,3 | 1,3 | 39 | 20 | 378 | 30 |
| 2 | 9,4 | 6,3 | 286 | 164 | 108 | 15 |
| 3 | 13,3 | 1,7 | 32 | 12 | 88 | 17 |
| 4 | 13,2 | 3,1 | 47 | 31 | 248 | 35 |
| 5 | 13,4 | 9,9 | 1163 | 339 | 68 | 22 |
| 6 | 2,5 | 0,6 | 43 | 26 | nr | 34 |
| 7 | 4 | 2,5 | nr | nr | nr | 25 |
| 8 | 12,4 | 4,4 | 154 | 93 | nr | 10 |
| 9 | 11,4 | 5,2 | 47 | 11 | 82 | 24 |
| 10 | 8,6 | 0,7 | 88 | 31 | nr | 32 |
| 11 | 5,8 | 3,2 | 52 | 30 | nr | 26 |
| 12 | 7,6 | 5,5 | 94 | 76 | nr | 36 |
| 13 | 9,7 | 6,5 | 194 | 160 | 312 | 6 |
| Referencias (rango) | 0,1 a 12,0a0,1 a 1,1b | 0,0 a 0,60 | 13 a 82 | 6 a 59 | 8 a 147 | 60 a 100 |
NR: no realizado.
La IH fue múltiple en los 12 pacientes con compromiso de los ejes corticotropo, tirotropo, somatotropo. Todos los varones, excepto uno (caso 2) tuvieron afectación del eje gonadotropo (tabla 3). Tres niños manifestaron nistagmo, uno al diagnóstico (caso 8) y 2 durante su seguimiento a los 2 y 6 meses de vida (casos 11 y 10). En 2 de ellos se constató hipoplasia de los nervios ópticos en el fondo de ojo.
Laboratorio hormonal e imágenes
| Paciente (#) | TSH (mU/l) | T4L (ng/dl) | ACTH (pg/ml) | Cortisol (μg/dl) | GH (ng/ml) | IGF-1 (ng/ml) | LH (U/l) | FSH (U/l) | T (ng/dl) | PRL (ng/ml) | Déficit de: | Diagnóstico | Imágenes |
| 1 | 10,6 | 0,6 | < 10 | 3a | 5,4a | ND | 0,06 | 0,1 | < 10 | 9 | TSH, ACTH, GH, LH/FSH | IH | Hipoplasia hipofisaria, agenesia tallo hipofisario. NHE |
| 2 | 5,43 | 0.3 | NR | 9a | 0,05 | ND | 9,5 | 7,9 | 440 | NR | TSH, ACTH, GH | IH | Hipoplasia hipofisaria |
| 3 | 8 | 0,53 | NR | < 1a | 0,7 | ND | < 0,05 | < 0,1 | – | 13,1 | TSH, ACTH, GH, LH/FSH, PRL | IH | Agenesia hipofisaria. NHE |
| 4 | 8,01 | 0,72 | NR | 3,6 | 0,54 | ND | 0,22 | 0,22 | < 10 | 22,1 | TSH, ACTH, GH, LH/FSH | IH | Hipoplasia hipofisaria. |
| 5 | 5,16 | 0,72 | 12a | < 1a | 3,1a | ND | <0,1 | < 0,1 | < 10 | 41 | TSH, ACTH, GH, LH/FSH | IH | Hipoplasia hipofisaria. NHE |
| 6 | <0.01 | < 0,02 | NR | < 1 | < 0,05 | ND | < 0,1 | < 0,1 | – | 0,5 | TSH, ACTH, GH, LH/FSH, PRL | IH | Agenesia hipofisaria y de tallo hipofisario |
| 7 | 6,86 | 0,5 | <10a | < 1a | 0,15a | ND | < 0,1 | 0,37 | – | < 1 | TSH, ACTH, GH, LH/FSH, PRL | IH | Tallo filiforme |
| 8 | 9,86 | 0,62 | 19 | < 1a | 4,6a | 16 | 0,71 | 2,2 | < 10 | NR | TSH, ACTH, GH, LH/FSH | IH | Hipoplasia hipofisaria y de tallo hipofisario. Ausencia señal neurohipófisis |
| 9 | 5,92 | 0,72 | < 10a | < 1 | 1,9 | 22 | < 0,05 | < 0,1 | < 10 | 144 | TSH, ACTH, GH, LH/FSH | IH | Hipoplasia hipofisaria, disrupción del tallo, NHE |
| 10 | 8,96 | 0,6 | 13 | <1 | 0,78b | ND | < 0,01 | 0,16 | < 10 | 28,7 | TSH, ACTH, GH, LH/FSH | IH | Agenesia de tallo hipofisario, ausencia señal neurohipófisis |
| 11 | 5 | 0,62 | NR | 1,1a | 1,5b | ND | 0,1 | 0,16 | < 10 | 144 | TSH, ACTH, GH, LH/FSH | IH | Hipoplasia hipofisaria |
| 12 | 6,7 | 0,48 | NR | 1,1a | 4,4a | ND | NR | NR | – | 1,2 | TSH, ACTH, GH, LH/FSH, PRL | IH | Hipoplasia hipofisaria |
| 13 | – | – | 982 | < 1a | – | – | – | – | – | – | Cortisol, aldosterona | IAP | Hipoplasia adrenal |
ACTH: adrenocorticotrofina; FSH: hormona folículo estimulante; GH: hormona de crecimiento; IGF-I: factor de crecimiento insulino símil tipo I; LH: hormona luteinizante; ND: no dosable; NHE: neurohipófisis ectópica; NR: no realizado; PRL: prolactina; PN: peso al nacer; T: testosterona; TSH: tirotropina; T4L: tiroxina libre.
Véanse valores normales en el texto (apartado «Métodos»).
En el paciente 13 el diagnóstico de IAP incluyó deficiencia tanto glucocorticoidea (tabla 3) como mineralocorticoidea, presentando un sodio plasmático de 120 mEq/l y un potasio de 8,5 mEq/l.
ImágenesLa ecografía hepática fue normal en 11 pacientes y con leve a moderada hepatomegalia en dos. La gammagrafía de excreción biliar con IDA se realizó en 3 pacientes y fue patológico en uno (falta de progresión del radioisótopo) a pesar de que finalmente las vías biliares fueron normales (falso positivo).
La RM de la región selar de los niños con IH demostró anomalías anatómicas de la hipófisis anterior (hipoplasia o agenesia) y/o del tallo hipotálamo hipofisario (adelgazamiento o agenesia) en todos los pacientes. La mayoría presentó alteraciones en la señal hiperintensa de la neurohipófisis (ectopia o ausencia) (tabla 3). En ningún caso se observaron malformaciones estructurales asociadas.
En el paciente con IAP la ecografía adrenal informó glándulas suprarrenales hipoplásicas.
EvoluciónPara el control de la glucemia durante el periodo neonatal, todos requirieron flujo de glucosa permanente por vía intravenosa o enteral continua entre 6 y 12,5mg/kg/min. Una vez instaurado el tratamiento con hidrocortisona, se logró disminuir gradualmente el flujo de glucosa hasta suspenderlo. El paciente con IAP y la mayoría de los pacientes con IH no repitieron episodios de hipoglucemias. En los restantes 3 pacientes se detectaron hipoglucemias con ayunos menores de 3 h, por lo que se debió iniciar tratamiento sustitutivo con hormona de crecimiento recombinante humana (rhGH) a los 2, 7 y 8 meses de vida para normalizar sus glucemias. Un paciente (caso 5) controló las glucemias con ayunos cortos hasta iniciar el tratamiento con hidrocortisona.
La hiperbilirrubinemia se resolvió en todos los pacientes al instaurar el tratamiento hormonal de remplazo apropiado (hidrocortisona tanto para los pacientes con IH como IAP sumado a levotiroxina para aquéllos con IH), excepto en un paciente en el cual la colestasis remitió espontáneamente previo al inicio de la terapia hormonal. En algunos casos, los niveles de bilirrubina disminuyeron al recibir tratamiento anticonvulsivante con fenobarbital sin llegar a normalizarse. El tiempo de persistencia de transaminasas elevadas fue variable, siendo el máximo periodo observado de 12 meses luego de haber iniciado el tratamiento sustitutivo.
DiscusiónEl abordaje diagnóstico de un lactante con ictericia prolongada con o sin colestasis, inicialmente se orienta hacia las patologías más prevalentes y graves como sepsis o atresia de vías biliares9,10. Existen pocas patologías que se presentan con ictericia neonatal acompañada de hipoglucemia, limitándose los diagnósticos diferenciales a sepsis, enfermedades metabólicas y endocrinopatías10,11. Dentro de este panorama diagnóstico, las enfermedades endocrinológicas constituyen una causa poco frecuente de ictericia prolongada en recién nacidos, afectando a menos del 3% de los neonatos con este cuadro. Sin embargo, esa probabilidad aumenta considerablemente cuando se suma la presencia de hipoglucemias recurrentes. Debido a la baja prevalencia de estas enfermedades, el diagnóstico de insuficiencia hormonal habitualmente es tardío y en ocasiones surge recién después de realizar una biopsia hepática12.
La demora en el diagnóstico de estos pacientes puede resultar en daño cerebral (retardo madurativo y/o epilepsia), causado principalmente por las hipoglucemias recurrentes y posiblemente agravado por un cuadro de hipotiroidismo central1,13, puede conducir a la fibrosis hepática progresiva por colestasis no resuelta o a la muerte por insuficiencia suprarrenal.
La sospecha diagnóstica se construye sobre la base de la confección de una minuciosa historia clínica, donde se detallan los antecedentes familiares y perinatales y se jerarquizan aspectos del examen físico sugestivos de alguna insuficiencia hormonal. Entre ellos, se destacan la hiperpigmentación cutaneomucosa en el caso de IAP y los signos asociados a posibles defectos de línea media (fisura labio alveolo palatina, nistagmus, microftalmos) o anomalías genitales (micropene, criptorquidia y/o microorquidismo) en los casos de IH14. A su vez, signos sutiles de hipoglucemia, como los antecedentes de hipotonía, hipotermia o succión débil, contribuyen a la orientación diagnóstica. Si bien no existen signos clínicos patognomónicos de hipotiroidismo central, la presencia de fontanela posterior permeable, hipotonía y somnolencia se describen en el hipotiroidismo primario y en el déficit de beta TSH15.
En el presente trabajo describimos las características clínicas y de laboratorio de 13 niños menores de 3 meses derivados a nuestro hospital por presentar ictericia prolongada e hipoglucemias que resultaron ser secundarias a patologías endocrinológicas. El niño con IAP se presentó con marcada hiperpigmentación cutaneomucosa, sin ninguna otra dismorfia. La mayoría de los varones con IH (7/8) presentaron algún grado de anomalía genital y en ellos se confirmó el hipogonadismo hipogonadotrófico. Los signos clínicos descriptos constituyeron una herramienta fundamental para orientar el diagnóstico de nuestros pacientes.
El patrón de afectación hepática secundario a una insuficiencia hormonal es variable y ha sido descrito con hiperbilirrubinemia de predominio indirecto, hiperbilirrubinemia de predominio indirecto de inicio que evoluciona a colestasis, colestasis de inicio o colestasis y hepatitis12. La fisiopatogenia se relaciona con una reducción de la síntesis primaria de ácidos biliares, un retraso en la síntesis y la maduración de enzimas de conjugación (bilirrubina uridina bifosfato glucurosiltransferasa) y también con alteración en la función de los canalículos biliares, que ocurren a una edad donde el metabolismo hepático y la excreción biliar son normalmente inmaduros16. Con frecuencia, se observa hiperbilirrubinemia de tipo indirecto en el hipotiroidismo congénito primario y ha sido descrito en el hipotiroidismo central por déficit de β-TSH15,17. En el caso de la colestasis, se propone al hipocortisolismo, tanto aislado como asociado a otras deficiencias de hormonas hipofisarias como el responsable de esta, apoyado por evidencias experimentales in vitro e in vivo donde se ha demostrado que los glucocorticoides influyen en la formación de bilis y favorecen la recirculación enterohepática de las sales biliares16,18,19. No existe un consenso claro sobre el papel que desempeñan las demás hormonas adenohipofisarias (GH u hormonas sexuales) en la patogenia de la colestasis neonatal en niños con IH. Publicaciones previas sugieren que algunas enzimas hepáticas son dependientes de GH, entre ellas la glucoquinasa ornitina descarboxilasa. Otros estudios experimentales demostraron que la hormona de crecimiento modula la biosíntesis y la secreción de los ácidos biliares20–22. Desde el punto de vista clínico, probablemente sea la deficiencia simultánea de varias hormonas la que contribuye a retrasar la maduración del mecanismo de transporte hepático de los ácidos biliares.
Si bien no existe una alteración bioquímica de disfunción hepática que sea patognomónica de insuficiencia hormonal, algunos autores han intentado identificar un patrón bioquímico que lleve a sospechar una causa endocrinológica.
El nivel de GGT suele ser normal o levemente elevado en los casos de IH, transformándolo en una herramienta útil para discernir si se trata de una patología extrahepática o si es primaria del hígado en caso de estar moderadamente aumentada23. Ellaway et al.24 enfatizan que el diagnóstico de hipopituitarismo congénito constituye un diagnóstico diferencial primordial en cualquier niño con colestasis y GGT normal o cercana a lo normal. Sin embargo, también han sido descritos casos de colestasis secundaria a IH con GGT aumentada, situación que se presentó en nuestra serie con 3 pacientes, confirmando que aún no existe un patrón bioquímico característico de disfunción hepática secundaria a una deficiencia hormonal. De acuerdo con la literatura, la histología de las biopsias hepáticas realizadas en niños con hepatitis colestática de causa endocrinológica fue informada en su mayoría como «hepatitis por células gigantes» y en algunos casos de mayor tiempo de evolución como cirrosis hepática con hipertensión portal12,16,24. En todos nuestros pacientes con colestasis el diagnóstico endocrinológico precedió a la toma de biopsia hepática y en vista que el tratamiento hormonal revirtió las manifestaciones hepáticas no hubo necesidad de efectuar la biopsia hepática. La colestasis se resuelve rápidamente en respuesta al tratamiento hormonal instaurado en el periodo neonatal. A la inversa, el retraso en el diagnóstico prolonga la colestasis y puede conducir a cirrosis e hipertensión portal16,24,25. Excepcionalmente, algunos curan espontáneamente previo al remplazo hormonal, como ocurrió en un paciente de nuestra cohorte.
La fisiopatología de la hipoglucemia secundaria a deficiencia de GH y/o cortisol involucra una inadecuada gluconeogénesis, un aumento de la utilización de glucosa por insuficiente inhibición de los efectos insulínicos y una disminución de los sustratos precursores para la gluconeogénesis.
Las hipoglucemias generalmente son graves y ocurren en los primeros días de vida; sin embargo, episodios menos severos pueden no manifestarse hasta que se presenta una situación de estrés que pone en evidencia la falta de mecanismos hormonales contrarreguladores rápidos, como son la GH y el cortisol.
El diagnóstico de IH se confirma mediante la determinación sérica de cortisol, ACTH, GH en hipoglucemia y del resto de las hormonas (TSH, T4, prolactina, gonadotrofinas) en condiciones basales y/o mediante pruebas funcionales dependiendo de la edad del paciente. Las determinaciones de GH y cortisol durante un episodio de hipoglucemia tienen un valor diagnóstico inestimable y evitan las pruebas de estímulo farmacológico.
En la IH el sector hormonal comprometido puede ser variable, no sólo en relación al tiempo de comienzo sino también en la severidad de las deficiencias hormonales encontradas26. En nuestra cohorte, todos los niños tuvieron IH múltiple con afectación simultánea de la mayoría de los ejes adenohipofisarios evaluados desde el momento de su presentación.
Una vez confirmado el diagnóstico de IH, la RM de cerebro provee el correlato de la alteración anatómica de la región selar y supraselar asociado al cuadro de deficiencia hormonal e identifica anomalías estructurales asociadas26. Para algunas series, la displasia septoóptica constituye la causa más frecuente de IH congénita. En nuestra serie sólo se diagnosticó en 2/12 pacientes27.
Si bien en ocasiones los episodios de hipoglucemia y la colestasis pueden ocurrir en forma asincrónica, la posibilidad de asociar ambos signos clínicos orienta de manera más firme a un trastorno hormonal subyacente.
En todos nuestros pacientes se logró un diagnóstico certero y oportuno previo a que ocurrieran alteraciones mórbidas severas, lesiones hepáticas irreversibles o muerte.
En conclusión, frente a un lactante con ictericia prolongada e hipoglucemias se deben descartar la insuficiencia hipofisaria y la insuficiencia adrenal primaria.
Creemos que esta descripción del curso de la colestasis neonatal asociada a hipoglucemias alertará al pediatra sobre la posible existencia de hipopituitarismo o insuficiencia adrenal en recién nacidos y lactantes, considerándolas entre los diagnósticos diferenciales en forma precoz, evitando estudios invasivos hepáticos innecesarios y disminuyendo la morbimortalidad inherente a estas patologías que tienen tratamiento específico.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
