La atrofia muscular espinal tipo i (AME tipo i, OMIM #253300) es una enfermedad neuromuscular causada por la degeneración de las neuronas motoras alfa de la médula espinal y cuyas manifestaciones clínicas son hipotonía, debilidad muscular progresiva y dificultad respiratoria. Tras la fibrosis quística, la AME es el segundo trastorno autosómico recesivo más frecuente, con una incidencia estimada de 1/6.000 a 1/10.000 nacimientos, y una frecuencia de portadores de 1/40-1/601,2. La AME es considerada una de las principales causas hereditarias de mortalidad infantil, por lo que el consejo genético y la detección de portadores en las familias es de gran importancia. Actualmente, sigue sin haber un tratamiento efectivo, aunque las investigaciones realizadas con ratones knockout dan resultados prometedores3,4.
Los genes de la supervivencia de la neurona motriz (SMN) están localizados en el cromosoma 5q13.3. El gen SMN1, telomérico, se transcribe correctamente produciendo una gran cantidad de proteína SMN estable (SMNFL). El gen SMN2, centromérico, difiere de SMN1 en el nucleótido 840 C>T que introduce un sitio nuevo de splicing, produce una pequeña cantidad de proteína SMNFL y una cantidad variable de proteína inestable que no contiene el exón 7 (SMNΔ7). El 95-99% de los pacientes de AME carecen de ambas copias del gen SMN1 debido a una deleción en homocigosis o una conversión génica de SMN1 en SMN25,6.
Se distinguen 4 tipos de AME, dependiendo de la edad de aparición de los síntomas, de la máxima actividad muscular conseguida y de la supervivencia lograda por el paciente: AME tipo i o enfermedad de Werdnig-Hoffman, que es la más severa, causando mortalidad infantil; tipo ii o AME infantil crónica (OMIM #253550); tipo iii (enfermedad de Wohlfart-Kugelberg-Welander, OMIM #253400) o AME juvenil, y tipo iv (OMIM #271150), que comienza en la edad adulta7,8.
Las pruebas genéticas actuales, tales como la Multiplex ligation-dependent probe amplification (MLPA) o la PCR en tiempo real, nos permiten conocer el número de copias de SMN1 de una manera rápida y fiable. La ausencia del exón 7 del gen SMN1 confirma el diagnóstico de AME. Estos análisis semicuantitativos alcanzan una sensibilidad del 95% y una especificidad del 100%, y mejoran la sensibilidad del diagnóstico hasta en un 98%9,10.
Se describe el caso de un paciente de sexo femenino de 3 meses y 25 días, que fue llevado al Servicio de Urgencias de Pediatría por rechazo alimentario. Nacida de embarazo a término y parto vaginal en posición cefálica a las 40 semanas y 4 días. A las 36-48h de vida la paciente presentó una pérdida de peso del 9,5% junto con leves signos de deshidratación. La paciente no realizaba una succión efectiva del seno materno, por lo que se pautó lactancia artificial con jeringa.
A los 2 meses y 5 días, la paciente acudió a urgencias por rechazo alimentario. A los 3 meses, por infección respiratoria. En este último episodio, en el examen físico se observó hipotonía en el tronco y en los 4 miembros, con arreflexia osteo-tendinosa, tórax campaniforme y resto anodino. Para encauzar el diagnóstico, se solicitaron pruebas complementarias: electromiografía (EMG) y resonancia magnética cerebral y de médula completa. El resultado de la EMG fue compatible con el diagnóstico de AME, observándose fibrilaciones y duración aumentada de los potenciales de acción de la unidad motora. Las resonancias no revelaron alteraciones significativas.
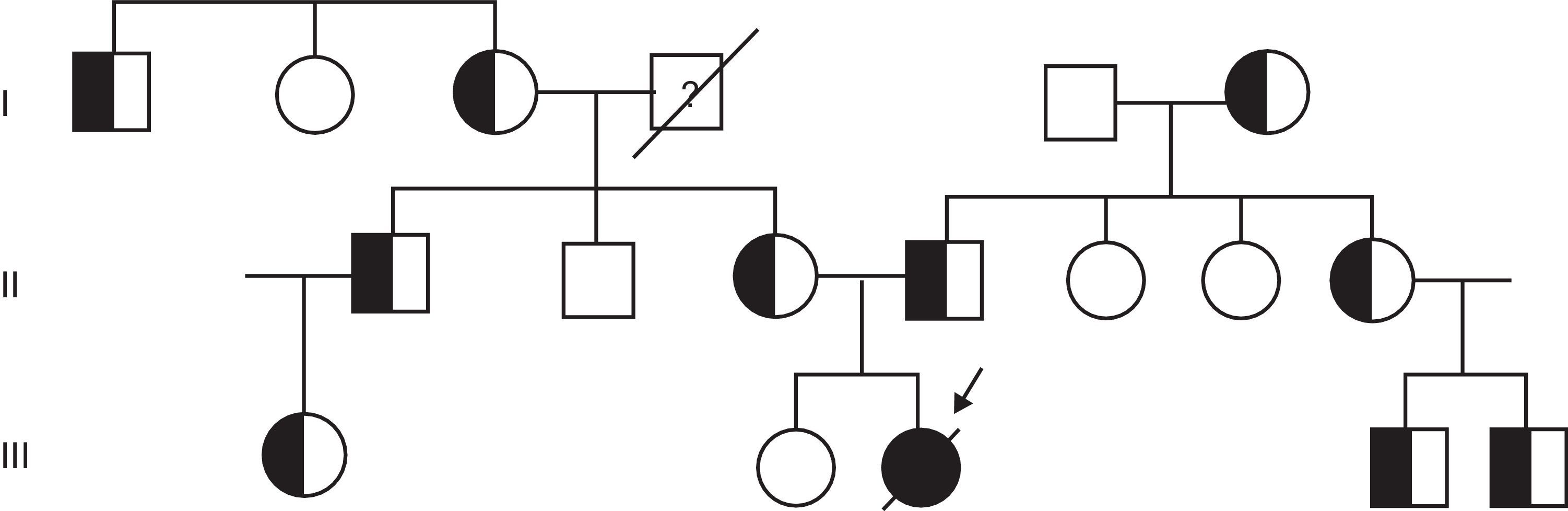
Tras estos hallazgos, se solicitó el estudio genético, que confirmó la sospecha diagnóstica de AME tipo i, informándose a la familia de la deleción en homocigosis de los exones 7 y 8 del gen SMN1 que presentaba la paciente y de la deleción en heterocigosis de ambos progenitores (fig. 1). Durante los meses siguientes al diagnóstico genético, la paciente fue ingresada en reiteradas ocasiones por afecciones respiratorias hasta su fallecimiento a los 8 meses y 26 días de edad.
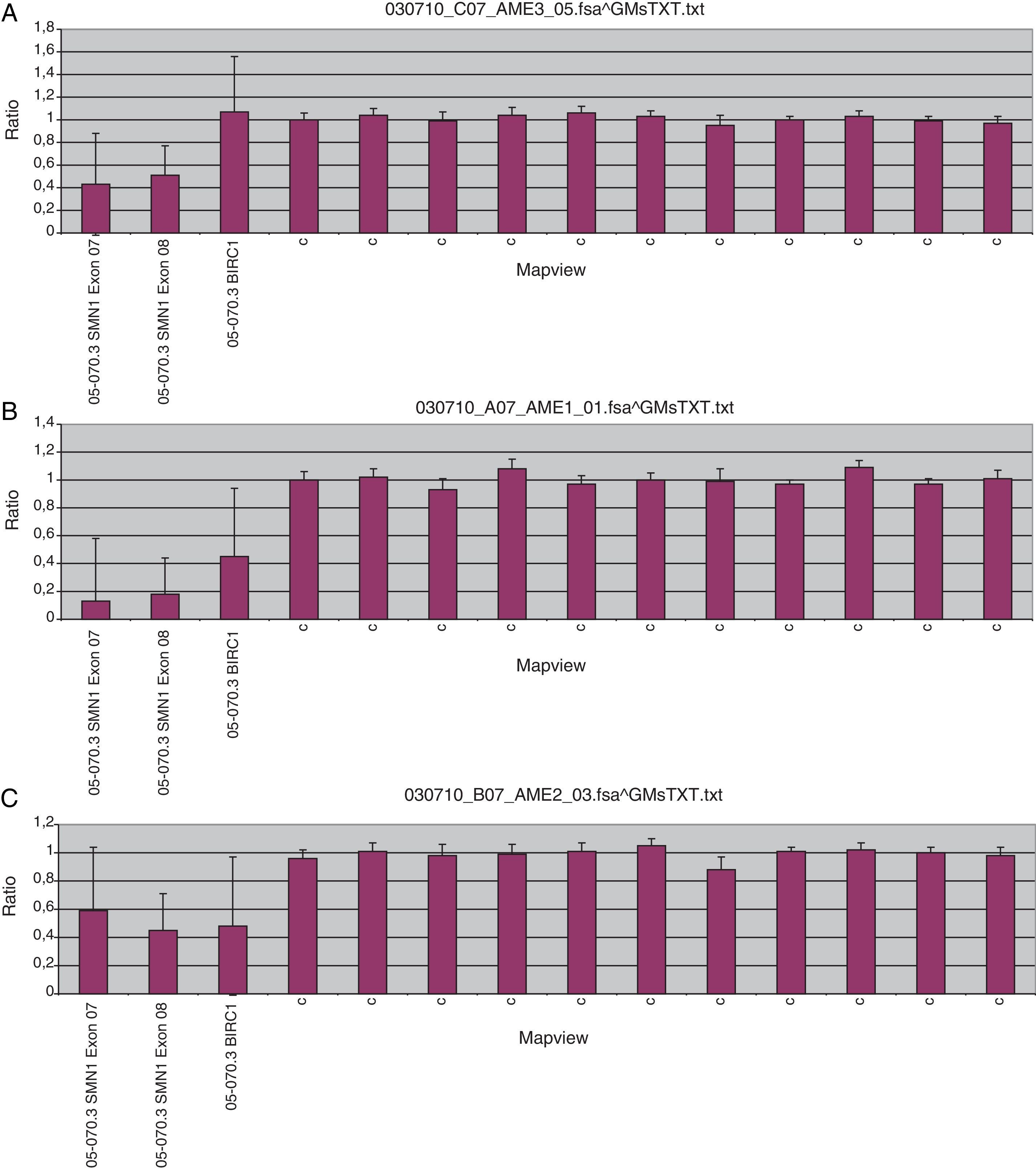
El histograma (A) muestra el resultado del análisis mediante MLPA del ADN materno, el histograma (B) muestra el resultado del caso índice y el histograma (C) muestra el resultado del ADN paterno. En el caso índice (B) se observa la pérdida de los exones 7 y 8 del gen SMN1, mientras que en el caso materno (A) se observa una pérdida del 50% de la dosis génica en los exones 7 y 8 (correspondientes a una sola copia del gen SMN1), mientras que en el ADN paterno (C) además se observa pérdida de dosis génica en el gen BIRC, un gen adyacente a SMN1. Resultados obtenidos a partir del kit P060-A2 SMA carrier (MRC-Holland).
La deleción se detectó mediante el kit P060-A2 SMA Carrier (MRC-Holland) y, a raíz del hallazgo de la mutación, se procedió al estudio de la misma en el resto de los familiares (fig. 2). Se realizaron un total de 17 análisis genéticos, encontrándose un total de 10 portadores. La familia materna era portadora de la deleción del exón 7 y 8 del gen SMN1 y la paterna era portadora de esta mutación y además de la deleción del gen BIRC, gen que colocaliza con el SMN1. El estudio de la AME tipo i mediante MLPA resultó un método rápido y fiable para la confirmación del diagnóstico y la identificación de portadores.