


Introducción
El déficit de acetoacetil-CoA tiolasa mitocondrial (T2) (OMIM 203750) recibe varios nombres, como los de déficit de betacetotiolasa, 3-oxotiolasa o aciduria alfametilacetoacética, si bien el más correcto es el mencionado en primer lugar. Se trata de una entidad englobada dentro de los errores innatos del metabolismo, con herencia autosómica recesiva y penetrancia variable que afecta al metabolismo de los cuerpos cetónicos y al catabolismo de la isoleucina1.
Clínicamente, se caracteriza por episodios agudos de cetoacidosis acompañados en ocasiones por pérdida de conciencia. Estos episodios se desencadenan por situaciones que cursan con aumento del metabolismo basal, tales como infecciones intercurrentes, crisis febriles o situaciones de ayuno prolongado.
Actualmente se desconoce con exactitud la prevalencia de esta enfermedad. Existen unos 40 casos publicados y, al menos, otros 20 pacientes están documentados.
La edad de presentación de esta entidad varía entre el primer año de vida y la adolescencia, y es excepcional su aparición antes de los seis meses. A continuación, presentamos un caso de déficit de betacetotiolasa con inicio en el período neonatal.
Caso clínico
Recién nacido de peso adecuado para la edad estacional (PAEG) que el cuarto día de vida presenta empeoramiento del estado general, polipnea con respiración tipo Kusmaull, letargia y rigidez generalizada. No existen antecedentes familiares de interés. En los antecedentes obstétricos destaca la colonización vaginal materna por estreptococo grupo B en el momento del parto, con tratamiento intraparto correcto. En ese momento presenta una pérdida máxima de peso del 13 % (peso: 2,7 kg), percentil 3-10, talla: 50 cm (P50).
En la exploración física destaca un grado de conciencia estuporoso, aspecto deshidratado, palidez generalizada con piel parcheada, irritabilidad ante estímulos mínimos y signos de distrés respiratorio, manteniendo saturación de oxígeno del 98 % con FiO2 0,21.
Las exploraciones complementarias al ingreso mostraban una acidosis metabólica grave (pH: 7,20; pCO2: 9 mmHg; HCO3: 3,5 mmol/l; BE: 21,8 mmol/l), con cetonemia intensa (> 6 mmol/l), hipernatremia moderada (Na: 156 mEq/l), hipopotasemia (K: 2,7 mEq/l) y aumento del anión GAP. La glucemia, el ácido láctico y el amonio fueron normales. La tira reactiva de orina mostraba proteinuria y cetonuria intensas. El resto de exploraciones complementarias efectuadas a su ingreso (hemograma, proteína C reactiva, cultivos seriados, radiografía de tórax, ecocardiografía y ecografía cerebral) no mostraron ninguna alteración.
Se procede a la reposición hidroelectrolítica y corrección de la acidosis con suero fisiológico y bicarbonato 1 molar. Se consigue una mejoría clínica y analítica notable en 24 h.
Realizamos recogida de muestras de sangre y orina en fase de descompensación clínica, para estudio de detección de metabolopatía. Se mantuvo a dieta absoluta durante tres días con fluidoterapia intravenosa, con aportes de glucosa de 7 mg/kg/min y administración de suplementos: L-carnitina, biotina y vitamina B12 por vía oral. Posteriormente, se procedió a la nutrición enteral con bajo contenido proteico (1,5-2 g/kg/día).
El estudio de ácidos orgánicos en orina por espectrometría de masas reveló un aumento del ácido 2-metil-3-OH-butírico, del ácido acetoacético y del 3-OH-isovalérico. Las concentraciones de tiglicina fueron indetectables. En una fase posterior de estabilidad clínica, se realizó una biopsia de piel para estudio de la actividad enzimática en linfocitos activados, y resultó un déficit total en la actividad de la acetoacetil-CoA tiolasa, con actividad residual indetectable (tabla 1). El estudio se efectuó en el Centro de Diagnóstico de Enfermedades Moleculares, Departamento de Biología Molecular, Universidad Autónoma de Madrid.

Actualmente, en un año y medio de vida, ha presentado tan sólo un episodio de descompensación cetoacidótica debido a una gastroenteritis aguda que le imposibilitaba el aporte adecuado de hidratos de carbono por vía oral, por lo que precisó fluidoterapia intravenosa durante dos días. En la actualidad, sigue una dieta con bajo contenido proteico (2 g/kg/día) y suplementos con L-carnitina.
Discusión
Las enfermedades metabólicas hereditarias suponen el 5 % de los ingresos pediátricos por urgencias en un hospital. Dado que pueden llegar a causar la muerte del paciente, es importante su diagnóstico de forma precoz2.
El déficit de T2, clínicamente se manifiesta como episodios cetoacidóticos intermitentes. Sin embargo, no existen síntomas clínicos entre cada crisis. Los episodios de cetoacidosis son normalmente graves, con descompensaciones asociadas al estrés metabólico y, a veces, se acompañan de letargia o incluso coma3,4. Algunos pacientes pueden presentar deterioro neuronal tras estos episodios. Aproximadamente dos tercios de los casos se presentan con alteración del estado de conciencia. En cambio, la hipoglucemia e hiperamoniemia son poco frecuentes. Se han descrito casos asintomáticos detectados durante el estudio familiar de un caso probando5. En nuestro paciente, la pérdida de peso y la escasa ingesta de los primeros días de vida pudieron ser los desencadenantes del inicio de su metabolopatía.
La acetoacetil-CoA tiolasa mitocondrial cataliza la transferencia reversible de moléculas de acetil-CoA, obtenidas en el proceso catabólico de la isoleucina, para formar acetoacetato CoA y CoA libre (fig. 1). En los tejidos extrahepáticos, la T2 interviene en la formación de acetil-CoA y energía. En el hígado, aporta acetoacetato CoA para la cetogénesis. Puede considerarse a la T2 como la primera enzima de la cetogénesis y la última que interviene en la cetolisis5. La falta de esta enzima implica un aumento de acetil-CoA, empleado como sustrato para la síntesis de 3-HMG-CoA en el hígado, que supone un aumento de 3-OH-butirato y acetoacetato1.
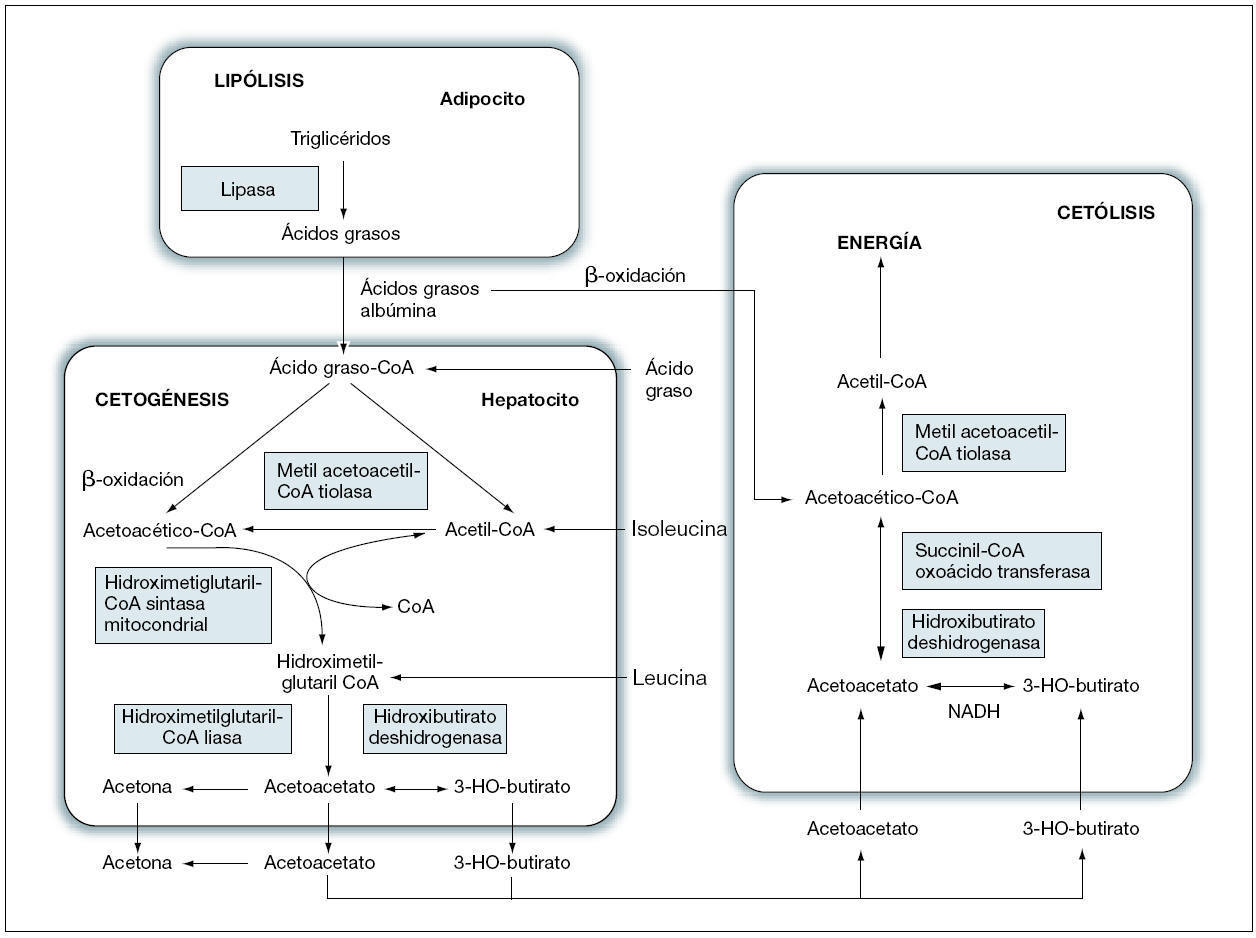
Figura 1. Vías de los cuerpos cetónicos.
Debe sospecharse la existencia de déficit de T2 en los casos en los que exista un aumento de la eliminación por orina de metabolitos de la isoleucina, 2-methyl-3-hydroxibutirato, tiglicina y 2-metilacetoacetato, si bien la ausencia de estos dos últimos no excluye el diagnóstico. Una situación de ayuno prolongado pone en evidencia una hipercetonemia intensa. En algunas ocasiones, la sobrecarga de la isoleucina puede ser útil para el diagnóstico5,6. El diagnóstico definitivo se realiza mediante la demostración del déficit enzimático en fibroblastos de la piel7. Fukao et al8 han identificado 24 mutaciones en 18 familias con déficit de T2.
Con respecto al inicio de este raro defecto enzimático, el primer episodio de cetoacidosis se presenta normalmente entre los 5 meses y los 2 años. La presentación neonatal es excepcional6. Un estudio reciente determinó los fenotipos y la forma de comienzo de 26 pacientes con déficit de T2 diagnosticados mediante análisis genético, y tan sólo recoge un caso con inicio en período neonatal9. En este caso, el paciente era de raza caucásica y presentó el primer episodio de cetoacidosis al tercer día de vida. Se demostró una actividad residual nula de la T2, como en el caso de nuestro paciente.
En los casos en los que el inicio se hace en período neonatal, es obligado hacer el diagnóstico diferencial con el déficit de succinil-CoA: 3-cetoácido-CoA transferasa (SCOT). Este trastorno se manifiesta con crisis recurrentes de cetoacidosis, y permanecen sin clínica en períodos intercrisis. A diferencia del déficit de acetoacetil-CoA tiolasa, no se pueden detectar ácidos orgánicos en orina. La cetonemia permanente es la característica más prominente en el déficit de SCOT. Aproximadamente la mitad de los pacientes con déficit de SCOT aparecen en período neonatal6.
Las secuelas clínicas se pueden evitar con un diagnóstico precoz y tratamiento apropiado de la cetoacidosis. El tratamiento no está consensuado, pero se aconseja la restricción proteica en la dieta, con aportes máximos entre 1,5-2 g/kg/día, para prevenir el potencial daño de la acumulación de los metabolitos de la isoleucina, que serían los causantes de la clínica neurológica durante las descompensaciones, y el riesgo de secuelas posteriores. Igualmente, una dieta rica en grasas induce la cetogénesis, por lo que se debe evitar. Estas recomendaciones se deben aplicar durante toda la vida del paciente6.
Asimismo, debe evitarse el ayuno prolongado, incluso valorar la administración profiláctica de glucosa en situaciones de aumento del catabolismo basal del organismo, como infecciones, fiebre o vómitos. Se recomienda realizar el control de cuerpos cetónicos en orina para conocer el grado de cetonemia en el organismo6. Se ha demostrado que las concentraciones de carnitina descienden en algunos pacientes con déficit de T210,11 por lo que se deben considerar suplementos con L-carnitina (50-200 mg/kg/día).
Para el diagnóstico de las metabolopatías en general se requiere que el pediatra esté alerta y familiarizado con los síntomas de presentación y en gran medida del apoyo de exámenes de laboratorio11. La importancia de su tratamiento precoz radica en la posibilidad de prevenir graves secuelas mentales y físicas y, en muchos casos, la muerte.
Correspondencia: Dra. I. Cubillo Serna.
Francisco Sarmiento, 13 4.º B. 09005 Burgos. España.
Correo electrónico: cibercubi@hotmail.com.
Recibido en diciembre de 2006.
Aceptado para su publicación en julio de 2007.
