
Introducción
El déficit de 11β -hidroxilasa es la segunda causa más frecuente de hiperplasia suprarrenal congénita (HSC), con una incidencia de 1 entre 100.000 recién nacidos vivos, lo que supone el 5-8 % de los casos de HSC1-3. Esta enzima está codificada por el gen CYP11B1 (8q21-q22), cuya alteración se transmite de forma autosómica recesiva y conlleva manifestaciones clínicas variables4. Estas se deben a acumulación tanto de 11-desoxicorticosterona, causante de hipertensión arterial, como de andrógenos suprarrenales, responsables de virilización neonatal e hiperandrogenización posterior5.
Se presenta una paciente con pubarquia a los 7 años de edad, maduración ósea acelerada y pronóstico de talla final muy inferior a su talla genética. En espera del resultado genético, que confirmó la sospecha de déficit de 11β -hidroxilasa, se inició tratamiento con corticoides orales y análogos de hormona liberadora de hormona luteinizante (LHRH). Con objeto de mejorar la talla final, se asoció hormona de crecimiento (GH). Con dicha combinación terapéutica, mejoró significativamente su pronóstico de talla y a los 13 años y 6 meses de edad presentaba una talla acorde a su talla genética.
Observación clínica
Niña de 7 años y 2 meses que consultó por pubarquia y olor corporal apocrino de un año de evolución, sin desarrollo de otros caracteres sexuales secundarios. Exploración física: peso: 24,5 kg (P64); talla 124 cm (P75); presión arterial 100/60 mmHg (P50-75). Pubarquia II en labios mayores y leve hipertrofia de clítoris. No axilarquia ni telarquia. Escoliosis dorsal de concavidad derecha.
Antecedentes personales: embarazo normal. Cesárea por desproporción pélvico-cefálica, a término. Peso al nacimiento 4.080 g (P95), longitud 52 cm (P > 97). Escoliosis dorsal de 18° en tratamiento ortopédico con corsé desde los 6 años, sin otros antecedentes médico-quirúrgicos de interés.
Antecedentes familiares: padres sanos, no consanguíneos. Talla materna: 158 cm (P24); talla paterna: 178 cm (P69). Un hermano, varón de 9 años, sano. Sin antecedentes familiares de pubertad adelantada (menarquia materna a los 15 años).
Se realizó estudio analítico inicial:
Sistemáticos de sangre y orina: normales.
Hormonas tiroideas: normales.
Estudio hormonal basal: cortisol: 9 µg/dl; ACTH: 24 pg/ml; IGF-I: 195 ng/ml; IGF-BP3: 3,5 µg/ml; sulfato de deshidroepiandrosterona (SDHEA): 0,94 µg/ml; 17-hidroxiprogesterona (17-OHP): 1 ng/ml; testosterona: 46 ng/dl; androstendiona: 2,8 ng/ml; 11-desoxicortisol: 57 ng/ml.
Test de LHRH: prepuberal.
Picos tras estímulo con ACTH: 17-OHP: 18 ng/ml; 11-desoxicortisol: 74 ng/ml; androstendiona: 4,43 ng/ml; resto normal.
En el estudio radiológico se observó edad ósea de 11 años (Atlas Greulich-Pyle) para una edad cronológica de 7 años y 4 meses. Ecografía tiroidea normal. Ecografía abdominopélvica sin alteraciones; útero y ovarios prepuberales y suprarrenales normales.
No se encontraron mutaciones en el gen CYP21B, que codifica para la enzima esteroide 21-hidroxilasa, por lo que se realizó secuenciación del gen de la 11β -hidroxilasa, hallándose la mutación T248I en heterocigosis (ACC > TGG) en el exón 4 del gen CYP11B1. Se trata de una conversión entre el gen CYP11B1 y el CYP11B2 que produce un cambio de una treonina a una isoleucina. No se practicaron más estudios genéticos en la paciente ni en sus familiares.
A la espera del estudio genético, se inició tratamiento con hidrocortisona oral (11 mg/m2/día) y un análogo de LHRH intramuscular, la triptorelina (3,75 mg/28 días), dado que en ese momento el pronóstico de talla final (Bayley-Pinneau) era de 140,4 cm (P < 3), con una talla genética de 164,6 ± 7 cm (P46).
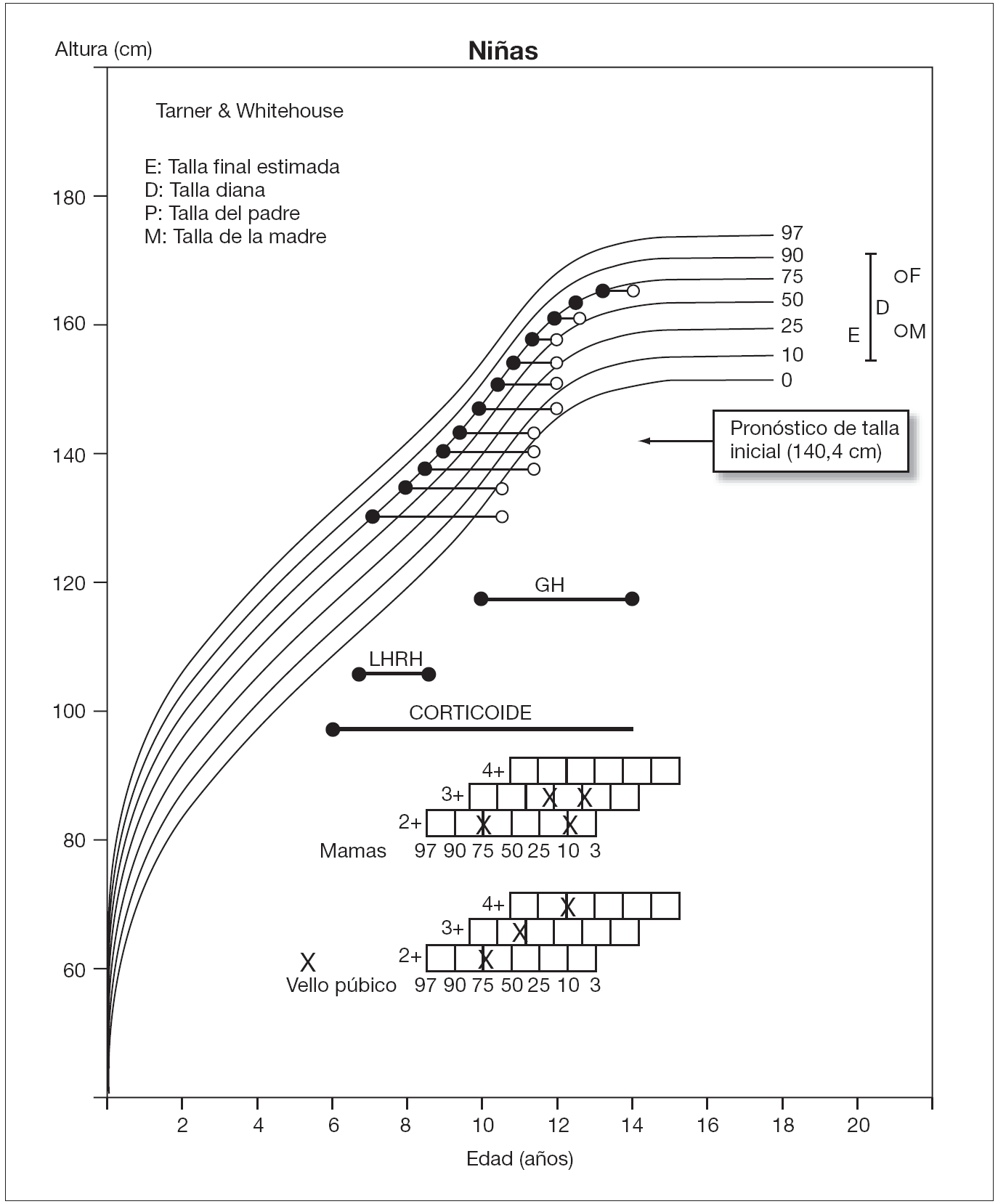
Se siguió clínica y analíticamente a la paciente con una periodicidad semestral (fig. 1), observando normalización de la función suprarrenal y mejoría progresiva del pronóstico de talla, sin aumento de los caracteres sexuales secundarios. Mantuvo una presión arterial dentro de la normalidad.
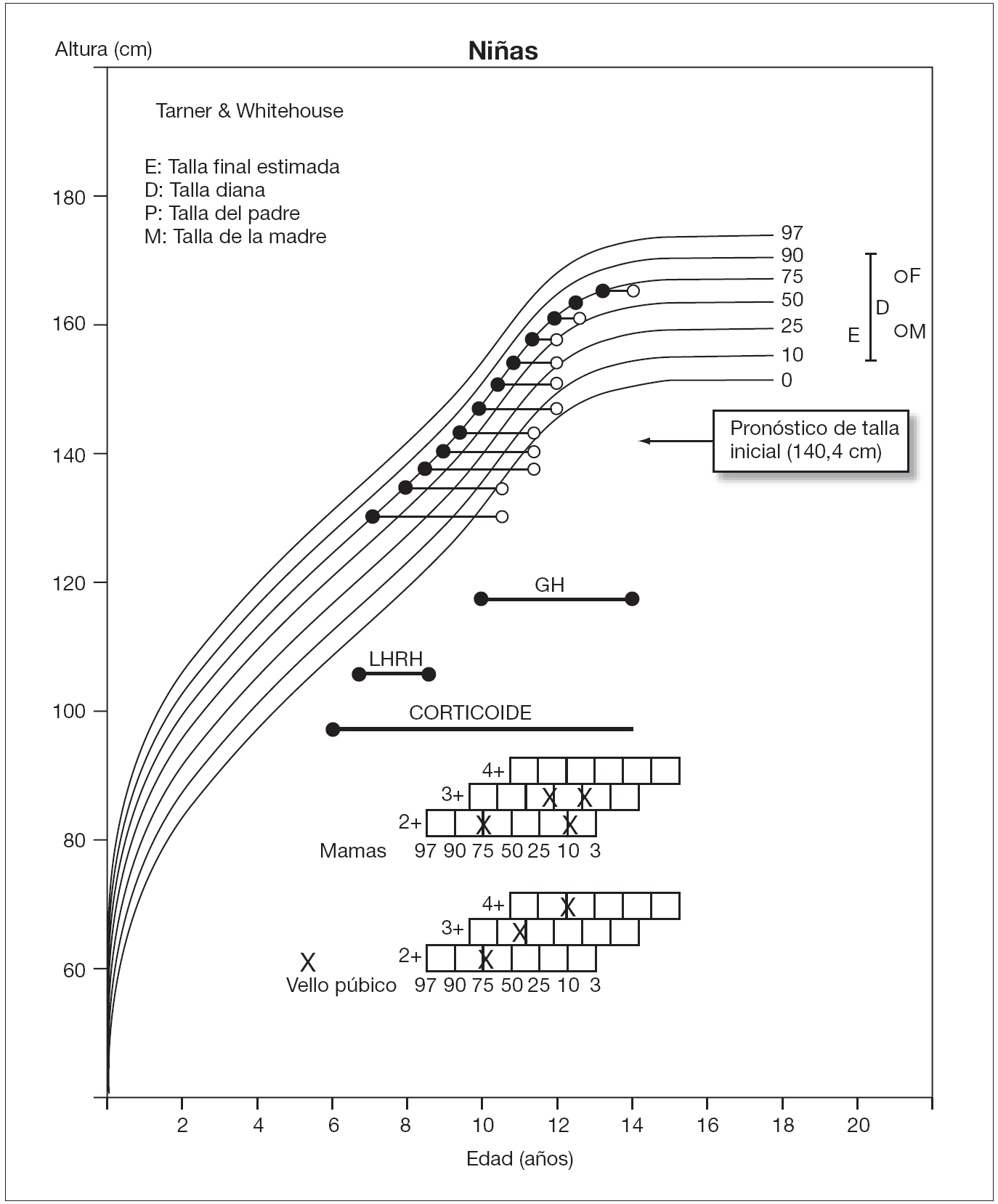
Figura 1. Evolución de talla (•) y edad ósea (°) durante el seguimiento de la paciente.
A los 9 años y 6 meses, con edad ósea de 12 años y pronóstico de talla final de 149,8 cm, se suspendió triptorelina y se mantuvo hidrocortisona (12 mg/m2/día).
No obstante, dado que la velocidad de crecimiento disminuyó progresivamente, se inició tratamiento con GH exógena (0,040 mg/kg/día) a los 10 años y 6 meses, con pubarquia II, telarquia incipiente, talla de 140 cm (P58), edad ósea de 12 años y pronóstico de talla final de 155,4 cm. Coincidiendo con el tratamiento administrado y el inicio puberal, experimentó una importante aceleración de la velocidad de crecimiento, sin avance significativo de maduración ósea.
A los 12 años y 11 meses, con desarrollo puberal en estadio III-IV de Tanner, talla de 153,5 cm (P32) y edad ósea de 13,5 años, su pronóstico de talla era de 157,6 cm, acorde a su talla genética. Se mantuvo tratamiento con hidrocortisona (13,8 mg/m2/día) y GH (0,036 mg/kg/día), sin observarse efectos adversos ni empeoramiento de su escoliosis. A la edad de 13 años y 6 meses, aún sin menarquia, se suspendió el tratamiento con GH, presentando una edad ósea de 14 años, talla de 157 cm y pronóstico de talla final de 159,7 cm (tabla 1). En ese momento, se programó cirugía correctora de su escoliosis, que tuvo lugar a los 14 años de edad, sin incidencias.
Discusión
La HSC es una de las enfermedades endocrinológicas más frecuentes. De herencia autosómica recesiva, se debe en más del 90 % de los casos al déficit de 21-hidroxilasa y en el 5-8 % al déficit de 11β -hidroxilasa1-3.
El déficit de 11β -hidroxilasa produce disminución en la producción de cortisol, incremento en la secreción de ACTH y acumulación de 11-desoxicorticosterona y andrógenos suprarrenales, que pueden producir hipertensión arterial e hiperandrogenismo, respectivamente6.
En el déficit severo la clínica es precoz, con virilización de los genitales externos de las niñas, que presentan gónadas y genitales internos femeninos normales, y con aumento del tamaño peneano en los niños. En otros casos, el hiperandrogenismo se manifiesta posnatalmente, como pubarquia precoz y rápido crecimiento somático, con aceleración de la maduración esquelética. Esta maduración somática puede desencadenar una pubertad precoz, la cual conlleva el cierre prematuro de las epífisis3,6. Todo ello explica que la talla baja final sea habitual en estos pacientes, siendo precisas estrategias terapéuticas que permitan mejorarla7. Adolescentes y adultos previamente asintomáticos pueden tener únicamente hirsutismo e irregularidades menstruales, como forma clínica más leve.
Aproximadamente dos tercios de los pacientes desarrollan hipertensión, habitualmente en la adolescencia y de intensidad generalmente moderada8. Es más frecuente en los casos de inicio clínico precoz3 y presumiblemente secundaria al exceso de 11-desoxicorticosterona, aunque existe escasa correlación entre sus niveles plasmáticos y la presión arterial8.
La clínica es más severa cuanto mayor es la pérdida de función enzimática que conlleva la alteración genética responsable9. En la paciente presentada no se registró hipertensión y las manifestaciones clínicas fueron de inicio posnatal y debidas al exceso androgénico, con adrenarquia precoz y maduración ósea acelerada. En el estudio genético realizado únicamente se halló una mutación en el exón 4 del gen CYP11B1, hasta ese momento no descrita, con conversión entre los genes CYP11B1 y CYP11B2, altamente homólogos y situados en tándem10,11. No se pudo realizar estudio genético a otros miembros de la familia.
La paciente, con afectación inicial del pronóstico de talla adulta, recibió corticoides, análogos de LHRH y GH y se monitorizó bioquímica y clínicamente, controlando presión arterial, signos de virilización, velocidad de crecimiento y maduración ósea, según las recomendaciones internacionales6.
Para corregir el déficit corticoideo existente y suprimir la hiperproducción de andrógenos, se administran corticoides a dosis sustitutivas5, estrategia conocida desde 195012. El exceso de corticoides interfiere en el crecimiento, de forma dosis-dependiente y multifactorial13, por lo que se ajustó periódicamente su dosificación, consiguiendo la normalización de los niveles de andrógenos (SDHEA, androstendiona y testosterona), inicialmente elevados.
Con objeto de frenar la maduración esquelética, se administraron análogos de LHRH hasta una edad ósea de 12 años (fig. 1). Con ellos se ha conseguido mejorar el pronóstico de talla final en ensayos previos, pero pueden reducir la velocidad de crecimiento14.
Se asoció GH al tratamiento para mejorar su pronóstico de talla final, significativamente comprometido respecto a su talla genética. Con las dosis recomendadas15 se ha constatado una mejoría significativa de la talla final en pacientes afectados de este déficit7,16, al igual que en el presente caso, que ha alcanzado su talla genética.
Por tanto, se puede concluir que la adición de GH y análogos de LHRH al tratamiento con corticoides constituye una estrategia terapéutica que puede mejorar significativamente la talla final de los pacientes con déficit de 11b -hidroxilasa, con pronóstico de talla adulta habitualmente comprometido.
Correspondencia: Dra. M. Orío Hernández.
Servicio de Endocrinología Pediátrica. Hospital Universitario Infantil La Paz.
P.º de la Castellana, 261. 28046 Madrid. España.
Correo electrónico: yeyaorio@yahoo.es
Recibido en mayo de 2007.
Aceptado para su publicación en mayo de 2007.
