
El síndrome de Dravet se caracteriza por una epilepsia resistente a fármacos de inicio en el primer año de vida con crisis con fiebre, y posterior evolución con déficit cognitivo y epilepsia con múltiples tipos de crisis. Habitualmente se diagnosticaba en torno a los 3-4 años de vida, pero el mejor conocimiento de las características de las crisis en los últimos años y el hallazgo de la alteración genética causal más frecuente han permitido adelantar el diagnóstico.
Pacientes y métodosSe presenta una serie de 14 niños diagnosticados de síndrome de Dravet o epilepsia del espectro Dravet. A las características de las crisis ocurridas durante el primer año de vida en estos pacientes, se aplican una serie de criterios de riesgo para ver si es posible hacer el diagnóstico en dicho período de tiempo. Resultados: en el 100% de los niños de esta serie se podía sospechar el diagnóstico en el primer año de vida aplicando estos criterios. Además, el 79% cumplían ya los criterios de riesgo en la primera crisis.
ConclusionesEs posible obtener un índice alto de sospecha de síndrome de Dravet en el primer año de vida. Es esencial, por tanto, la difusión de los criterios clínicos que permiten la sospecha diagnóstica temprana y la distinción de crisis febriles de otro origen, y el establecimiento de un protocolo de recogida de datos para las crisis con fiebre en el primer año de vida.
Dravet syndrome is a drug resistant epilepsy which starts in the first year of life with febrile seizures, followed by cognitive impairment and epilepsy with multiple seizure types. Diagnosis has been typically made at the age of three to four years, but earlier diagnosis is now possible as clinical features are better recognised and molecular diagnosis is available.
Patients and methodsWe studied a series of 14 children with Dravet syndrome or Dravet spectrum epilepsy. A screening test, developed by other authors to distinguish the febrile seizures in Dravet syndrome from febrile seizures from other origin, was applied to the clinical features of the seizures occurring during the first year of life in our patients.
ResultsClinical suspicion of Dravet spectrum epilepsy was possible in 100% of children in our series. Moreover, taking into consideration only the first seizure, 79% of patients scored sufficiently to detect Dravet syndrome.
ConclusionsDravet syndrome can be recognised during the first year of life. It is important that physicians are made aware of these clinical criteria capable to distinguish febrile seizures in Dravet syndrome from febrile seizures of other origin, and set up a protocol to collect appropriate data regarding febrile seizures occurring in the first year of life.
El síndrome de Dravet es una encefalopatía epiléptica caracterizada por la presencia de crisis predominantemente desencadenadas por fiebre durante el primer año de vida, en un lactante con un desarrollo normal, seguidas de epilepsia con distintos tipos de crisis a partir del segundo año1. La evolución es hacia una epilepsia farmacorresistente y un estancamiento en el desarrollo psicomotor a partir del segundo año de vida, que lleva finalmente a un déficit cognitivo entre moderado y severo.
En el año 2001 se descubrieron alteraciones en el gen SCN1A en 7 pacientes estudiados2 y desde entonces numerosos estudios confirman alteraciones en este gen en aproximadamente 80% de los pacientes con Dravet3–13. Actualmente se considera que existe un espectro clínico que incluye desde el extremo benigno de las convulsiones febriles plus hasta los cuadros de pronóstico más reservado como la epilepsia intratable de la niñez con crisis generalizadas tónico-clónicas14,15, la epilepsia multifocal severa del lactante9 o las formas clásica y borderline del síndrome de Dravet, ya que se encuentran alteraciones del gen SCN1A en pacientes con cualquiera de estos fenotipos. La tendencia actual es considerarlo un espectro, que en este artículo denominaremos espectro Dravet (ED).
El diagnóstico de síndrome de Dravet se ha realizado clásicamente hacia los tres o cuatro años de vida, momento en el que la aparición de distintos tipos de crisis y la evidencia del estancamiento en el desarrollo psicomotor hacían posible la confirmación clínica. Las crisis iniciales, especialmente si todas ocurrían con fiebre, eran consideradas convulsiones febriles de mayor o menor atipicidad, pero no se iniciaba tratamiento en la mayoría de los casos. En la actualidad se conocen mejor las características típicas de las crisis con fiebre propias del síndrome de Dravet y esto, junto con la posibilidad de obtener un estudio molecular del gen SCN1A, ha permitido adelantar el diagnóstico de muchos de estos niños. La detección temprana del síndrome es de enorme importancia, ya que solo si se pone el tratamiento adecuado y se evitan fármacos nocivos, es posible mejorar el control de las crisis. Además, la detección temprana permite iniciar un programa de estimulación cognitiva que puede ayudar en el desarrollo psicomotor de estos niños.
En este estudio se aplican, a una serie de pacientes con epilepsia dentro del ED, unos criterios que han demostrado alta sensibilidad y especificidad para distinguir las crisis con fiebre del primer año de vida del síndrome de Dravet de las crisis con fiebre de otro origen. El objetivo es detectar la edad a partir de la cual se puede, en cada caso, empezar un tratamiento farmacológico incisivo de las crisis, justificado por una alta probabilidad de estar ante el diagnóstico de síndrome de Dravet. Asimismo, se da a conocer a la comunidad de pediatras una forma de sospecha diagnóstica muy eficaz y precoz que redundará de forma muy positiva en el manejo de los niños con este tipo de epilepsia.
Pacientes y métodosSe realizó un estudio retrospectivo de los pacientes diagnosticados de síndrome de Dravet y su espectro seguidos en nuestra unidad. Los criterios de inclusión para el estudio fueron: la presencia de crisis con fiebre en el primer año de vida y la comprobación de alteración en el gen SCN1A. Se realizó una revisión detallada de las historias clínicas, se recogieron las características de las crisis con fiebre en el primer año de vida de cada paciente, y se aplicó un sistema de puntuación de dichas características que se ha demostrado que permite hacer el diagnóstico del síndrome de Dravet.
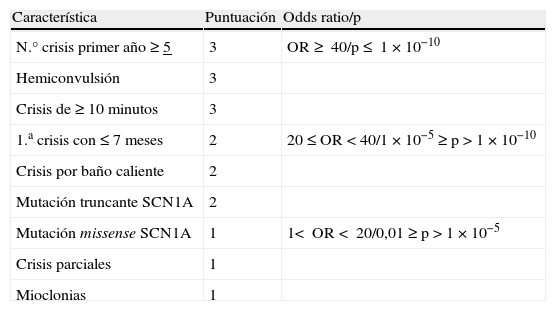
Las características se eligieron según lo propuesto por Hattori et al16 en su estudio de discriminación sobre 96 niños que tuvieron crisis con fiebre en el primer año de vida. Los dividieron en dos grupos según su evolución: 50 niños fueron diagnosticados de Dravet y 46 niños de crisis con fiebre en el primer año de vida de distinto origen. Estos autores, mediante regresión logística, calcularon la probabilidad que tenían los distintos rasgos de las crisis de predecir la aparición de un síndrome de Dravet. Obtuvieron una puntuación de riesgo del 1 al 3 según la odds ratio o la significación estadística asociadas a cada característica de riesgo capaz de diferenciar entre el grupo con SD y el grupo sin SD. Las puntuaciones y las características más discriminativas están en la tabla 1.
Características clínicas de las crisis con fiebre, puntuación según odds ratio y p para distinguir entre Dravet-no Dravet
| Característica | Puntuación | Odds ratio/p |
| N.° crisis primer año≥5 | 3 | OR≥ 40/p≤ 1×10−10 |
| Hemiconvulsión | 3 | |
| Crisis de≥10 minutos | 3 | |
| 1.a crisis con≤7 meses | 2 | 20≤OR<40/1×10−5≥p>1×10−10 |
| Crisis por baño caliente | 2 | |
| Mutación truncante SCN1A | 2 | |
| Mutación missense SCN1A | 1 | 1< OR< 20/0,01≥p>1×10−5 |
| Crisis parciales | 1 | |
| Mioclonias | 1 |
El punto de corte clínico para identificar el SD se estableció en ≥6 puntos, en caso de incluirse el baño típico japonés a elevada temperatura, o en ≥5 para niños en otras culturas. Nosotros hemos empleado este último. Si se añadía el estudio molecular, el punto de corte se establecía en ≥6 (sin baño caliente), y se incrementaban la especificidad de 0,880 a 0,900, con un valor predictivo positivo del 98,9%.
Se revisaron las historias clínicas de 18 pacientes con crisis con fiebre en el primer año de vida y alteración en el gen SCN1. Aplicamos la puntuación propuesta por Hattori et al a las características de las crisis con fiebre que estos niños padecieron en su primer año de vida. Se valoró la edad a la cual se podía hacer un diagnóstico clínico y se determinó la aportación del estudio genético como posible factor para adelantar la edad de diagnóstico.
ResultadosDe 18 pacientes evaluados, se seleccionaron 14 (seis mujeres y ocho varones) por constar con exactitud en la historia clínica los datos necesarios de las crisis en el primer año. En el estudio de Hattori et al, los 50 niños estaban diagnosticados de síndrome de Dravet. En nuestra serie, 8 niños tienen síndrome de Dravet clásico, 3 borderline, uno posiblemente convulsiones febriles plus y en los dos restantes no hay edad suficiente para definir bien el fenotipo. Por este motivo, al referirnos a nuestra serie, hablaremos de espectro Dravet y no de síndrome de Dravet.
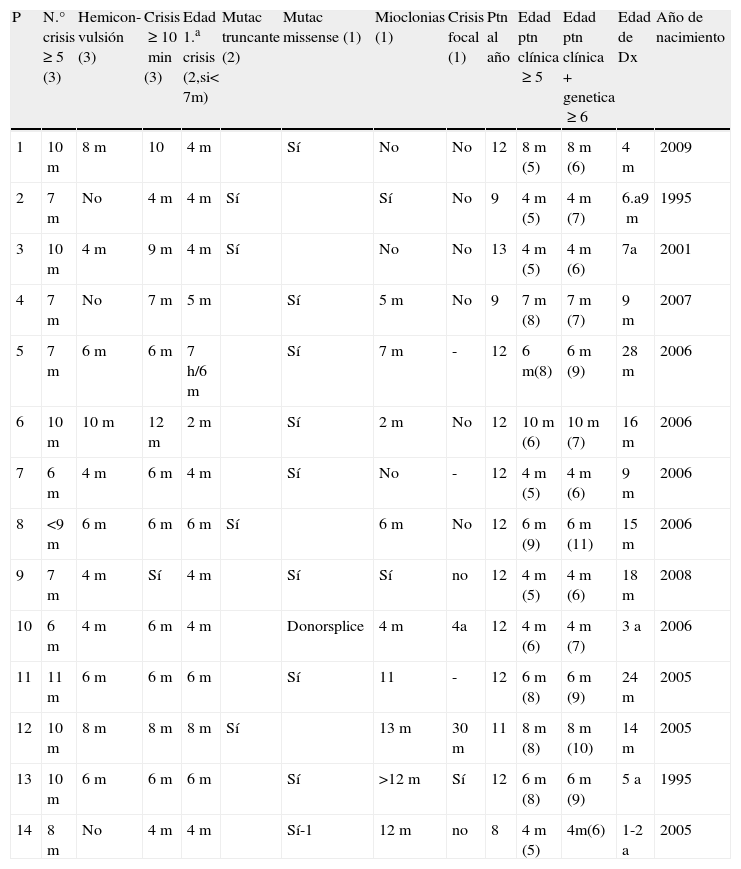
En la tabla 2 se presentan las características de las crisis y la edad de aparición, la puntuación que obtiene cada paciente en el primer año de vida según los criterios de Hattori et al16, la edad más temprana en la que se hubiera podido hacer el diagnóstico clínico, la edad a la que se habría adelantado el diagnóstico en caso de obtener el diagnóstico molecular, y asimismo se presenta la edad real a la que se realizó el diagnóstico de ED.
Edad de aparición de las características de las crisis en el primer año de vida, puntuación obtenida según los criterios de Hattori et al al año de vida, edad de cumplimiento de criterios necesarios para el diagnóstico, edad de diagnóstico
| P | N.° crisis ≥5 (3) | Hemicon-vulsión (3) | Crisis≥10min (3) | Edad 1.a crisis (2,si< 7m) | Mutac truncante (2) | Mutac missense (1) | Mioclonias (1) | Crisis focal (1) | Ptn al año | Edad ptn clínica≥5 | Edad ptn clínica+genetica≥6 | Edad de Dx | Año de nacimiento |
| 1 | 10m | 8m | 10 | 4m | Sí | No | No | 12 | 8m (5) | 8m (6) | 4m | 2009 | |
| 2 | 7m | No | 4m | 4m | Sí | Sí | No | 9 | 4m (5) | 4m (7) | 6.a9m | 1995 | |
| 3 | 10m | 4m | 9m | 4m | Sí | No | No | 13 | 4m (5) | 4m (6) | 7a | 2001 | |
| 4 | 7m | No | 7m | 5m | Sí | 5m | No | 9 | 7m (8) | 7m (7) | 9m | 2007 | |
| 5 | 7m | 6m | 6m | 7h/6m | Sí | 7m | - | 12 | 6 m(8) | 6m (9) | 28m | 2006 | |
| 6 | 10m | 10m | 12m | 2m | Sí | 2m | No | 12 | 10m (6) | 10m (7) | 16m | 2006 | |
| 7 | 6m | 4m | 6m | 4m | Sí | No | - | 12 | 4m (5) | 4m (6) | 9m | 2006 | |
| 8 | <9m | 6m | 6m | 6m | Sí | 6m | No | 12 | 6m (9) | 6m (11) | 15m | 2006 | |
| 9 | 7m | 4m | Sí | 4m | Sí | Sí | no | 12 | 4m (5) | 4m (6) | 18m | 2008 | |
| 10 | 6m | 4m | 6m | 4m | Donorsplice | 4m | 4a | 12 | 4m (6) | 4m (7) | 3 a | 2006 | |
| 11 | 11m | 6m | 6m | 6m | Sí | 11 | - | 12 | 6m (8) | 6m (9) | 24m | 2005 | |
| 12 | 10m | 8m | 8m | 8m | Sí | 13m | 30m | 11 | 8m (8) | 8m (10) | 14m | 2005 | |
| 13 | 10m | 6m | 6m | 6m | Sí | >12m | Sí | 12 | 6m (8) | 6m (9) | 5 a | 1995 | |
| 14 | 8m | No | 4m | 4m | Sí-1 | 12m | no | 8 | 4m (5) | 4m(6) | 1-2 a | 2005 |
Min: minutos; p: paciente; ptn: puntuación. Se muestra entre paréntesis la puntuación de cada característica.
Al año de vida, se podía hacer el diagnóstico clínico de ED en el 100% de los niños estudiados si consideramos la puntuación de Hattori et al (puntuación≥5), que permite distinguir el ED de las convulsiones con fiebre de otro origen en el primer año de vida con una sensibilidad del 0,957 y una especificidad del 0,880. El análisis genético no incrementó en esa edad el rendimiento diagnóstico, aunque confirmó la sospecha clínica, ya que en el estudio antes mencionado eleva la especificidad a 0,900, con un valor predictivo positivo del 98,9%.
Además, el 79% de los pacientes ya cumplían criterios clínicos suficientes para el diagnóstico de espectro del Dravet en la primera crisis según el modelo 4 de Hattori et al, que tiene una sensibilidad de 0,957 y una especificidad de 0,880. La mediana de edad más temprana de posible diagnóstico clínico es de 6 meses en esta serie para los criterios clínicos (punto de corte de 5) y la mediana de edad de la primera crisis es 4 meses. En ningún caso se hubiera adelantado el diagnóstico solicitando el estudio molecular, pero el hecho de encontrar mutaciones en el gen SCN1A en ese momento hubiera incrementado ligeramente la especificidad, disminuyendo la posibilidad de falsos negativos.
La mediana de meses transcurridos desde que los pacientes cumplían criterios de riesgo hasta que se realizó el diagnóstico fue de 12 meses, con un rango que varía desde 0 a 77 meses de retraso diagnóstico. Si separamos los pacientes diagnosticados antes del año 2002, entonces solo fueron 6 meses la mediana de retraso entre el momento en que el paciente cumplía criterios y el momento en que se hizo el diagnóstico. Todos los pacientes de esta serie fueron diagnosticados por especialista en Neuropediatría y, entre ellos, al menos 12 en consulta especializada de epilepsia. Esto indica que, en esta serie, el tiempo transcurrido desde que se podía sospechar el diagnóstico hasta que se hizo, fue razonablemente corto. No obstante, es de esperar que si el diagnóstico se intenta realizar por el pediatra en la primera crisis, se acorte aún más el tiempo y se puedan tratar los niños de forma más precoz.
DiscusiónEn este estudio se muestra cómo, empleando los criterios clínicos estudiados por Hattori et al16, es posible hacer un diagnóstico de epilepsia del ED muy temprano, con alta sensibilidad y especificidad, en niños que se presentan con crisis con fiebre en el primer año de vida. Este dato es de enorme relevancia para los pediatras, que son los primeros que reciben al niño que ha tenido una crisis con fiebre. El hecho de que en el 79% de los niños de esta serie se hubiera podido sospechar el diagnóstico recogiendo con precisión las características de la primera crisis, hace esencial la difusión de estos criterios y el establecimiento de un protocolo de recogida de datos para crisis con fiebre en el primer año de vida en Atención Primaria y Servicios de Urgencias Hospitalarias. El 100% de los niños de nuestra serie cumplían criterios suficientes para el diagnóstico dentro del primer año de vida. En la serie de Ragona et al13, el 75% de sus pacientes también tenían una puntuación que indicaba una elevada probabilidad de tener Dravet, confirmando la eficacia de estos criterios para detectar casos de forma precoz.
Las crisis con fiebre del espectro Dravet son comúnmente confundidas al inicio del cuadro con convulsiones febriles (CF), especialmente con convulsiones febriles atípicas. Las CF se consideran crisis ocasionales relacionadas con un desencadenante, la temperatura, que fácilmente provoca crisis entre los 6 meses y los 6 años en niños con propensión genética a padecerlas. Es un cuadro frecuente, afecta al 5% de la población, benigno, que cede con la edad y no precisa tratamiento salvo en situaciones excepcionales17. El síndrome de Dravet constituye el extremo contrario: es una epilepsia farmacorresistente, que evoluciona con déficit cognitivo y debe tratarse de forma contundente, ya que cursa con una propensión a padecer estatus epilépticos y tiene una tasa de muerte súbita mayor de la población general y mayor de la que se encuentra en las epilepsias en general. Parece, por tanto, relevante poder distinguir ambas entidades de forma temprana. Los criterios propuestos por Hattori et al16 discriminan muy bien y, aplicados a nuestra serie, permiten distinguir el Dravet de las CF en el primer año de vida. De hecho, en el 79% de nuestros casos se podía diagnosticar con la primera crisis, lo cual permite instaurar un tratamiento muy precoz. Los beneficios del tratamiento temprano respecto a un mayor control de las crisis, la disminución de la tasa de muerte súbita o una evolución cognitiva mejor se desconocen, ya que no hay todavía series con suficiente número de pacientes diagnosticados pronto. Es un reto que pasa por la identificación temprana del síndrome por los pediatras y la adecuada e inmediata derivación de los pacientes a servicios con experiencia
Los niños con epilepsias dentro del espectro del Dravet, además de tener crisis con fiebre, frecuentemente experimentan crisis durante infecciones más o menos banales con temperatura menor de 38° C o incluso estando apiréticos. Esta tendencia los hace especialmente propensos a tener crisis tras las vacunaciones. De hecho, lo que se consideraba un encefalopatía epiléptica posvacunal se ha constatado que es debida a mutaciones en este gen SCN1A, y por tanto, la vacunación no tiene una responsabilidad causal en la aparición de la encefalopatía epiléptica18. Se recomienda por tanto continuar con el calendario vacunal que no solo no es nocivo sino especialmente conveniente en estos niños, quienes tendrán más crisis si tienen más infecciones. Es también aconsejable cubrir el período en el que habitualmente se tienen reacciones febriles en cada tipo de vacuna pautando antipiréticos sistemáticamente y añadiendo o incrementando, según el caso, clobazam al tratamiento de base antiepiléptico durante esos días.
La confirmación con diagnóstico molecular del síndrome de Dravet no es estrictamente necesaria para poder realizar el diagnóstico en una primera instancia. En nuestra serie, ninguno de los pacientes hubiera obtenido antes el diagnóstico añadiendo, a la puntuación obtenida con criterios únicamente clínicos, la puntuación resultante del estudio de gen SCN1A (tabla 2). Sin embargo, la confirmación genética incrementa la especificidad, dando así seguridad cuando hace falta instaurar tratamiento antiepiléptico en politerapia y con fármacos no exentos de efectos secundarios. Es por tanto necesario hacer el estudio molecular de forma precoz en el curso de la enfermedad. La afectación de los canales de sodio voltaje dependientes obliga a tener mucha precaución para evitar determinados fármacos antiepilépticos que pueden empeorar las crisis. La mayoría de casos diagnosticados de forma tardía han recibido antiepilépticos como la carbamacepina, la oxcarbacepina, la lamotrigina o la vigabatrina, que empeoran la epilepsia en los pacientes del espectro Dravet19,20. Recientemente se ha encontrado también un empeoramiento de la epilepsia en el 30% de niños tratados con rufinamida21. También tiene efectos secundarios más severos la fenitoína22 y es conveniente evitar los barbitúricos a dosis elevadas, dos fármacos que con frecuencia se utilizan los intensivistas en casos de estatus epiléptico23. Por otra parte, hay fármacos, como el estiripentol, que han demostrado una eficacia en el control de las crisis clónicas en niños con Dravet y que deben ser considerados como tratamiento en estos niños, a pesar de ser fármacos poco empleados en otros tipos de epilepsias24. También se conoce que los estatus pueden responder bien al midazolam endovenoso25.
En un estudio retrospectivo de pacientes adultos, observaron que la evolución cognitiva variaba, pero la mayoría de los pacientes tenía un déficit cognitivo entre moderado y severo26. La estimulación temprana de los niños diagnosticados pronto es esencial, ya que hay un primer período de desarrollo casi normal, seguido de un estancamiento que puede ser menor en la medida en que el niño sea estimulado y aproveche mejor sus recursos en los primeros 2 años de vida. Los beneficios de esta atención temprana no están aún demostrados por no haber seguimiento suficiente, pero parece lógico que apoyar su aprendizaje, junto con un mejor control de las crisis, pueda favorecer un mejor desarrollo cognitivo posterior. En estudios recientes se sugiere que el estancamiento cognitivo no depende de la epilepsia únicamente, sino de factores intrínsecos a la enfermedad27. Si se afectan canales de sodio voltaje dependientes, es de suponer que las funciones cognitivas de alguna manera se puedan ver interferidas.
Con este estudio concluimos que el síndrome de Dravet puede ser diagnosticado en el primer año de vida, lo que redundará en su mejor tratamiento. El conocimiento por los pediatras, especialmente de Atención Primaria y Urgencias, de los criterios propuestos por Hattori et al, permiten muy bien sospechar que una convulsión febril pueda ser el inicio de un Dravet y no una convulsión febril típica benigna. Es esencial, por tanto, la difusión de estos criterios y el establecimiento de un protocolo de recogida de datos para las crisis con fiebre en el primer año de vida.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
